Свойства и функции белков — урок. Химия, 8–9 класс.
Денатурация — разрушение пространственной структуры белка.
Она происходит при нагревании белков, под действием радиоактивного излучения, некоторых химических веществ (кислот, щелочей, солей тяжёлых металлов). При денатурации белки изменяют свои свойства и теряют биологическую активность, несмотря на то, что их первичная структура сохраняется.
Рис. \(1\). Денатурация белка
Примером денатурации служит изменение яичного белка при нагревании.
Разложение при нагревании
При сильном нагревании белки горят. При этом образуются вещества со своеобразным запахом жжёных перьев. По запаху можно легко отличить шерстяные или шёлковые волокна от синтетических.
Цветные реакции
Присутствие белка в растворе можно обнаружить с помощью качественных реакций, в результате которых образуются окрашенные продукты.
Если к раствору белка добавить раствор щёлочи и несколько капель раствора соли меди(\(II\)), то появляется красно-фиолетовое окрашивание. Эта реакция называется биуретовой.
Рис. \(2\). Биуретовая реакция
Другая цветная реакция на белки — ксантопротеиновая. Для её проведения к раствору белка при нагревании надо добавить концентрированную азотную кислоту. Образуется жёлтый осадок. Если после охлаждения в пробирку прилить раствор щёлочи или концентрированный раствор аммиака, то появится оранжевое окрашивание.
Рис. \(3\). Ксантапротеиновая реакция
Функции белков
В каждом живом организме содержится большое количество белков, которые выполняют ряд важнейших функций.
Белки входят в состав цитоплазматической мембраны, цитоплазмы, органоидов и тем самым выполняют строительную функцию в живых организмах.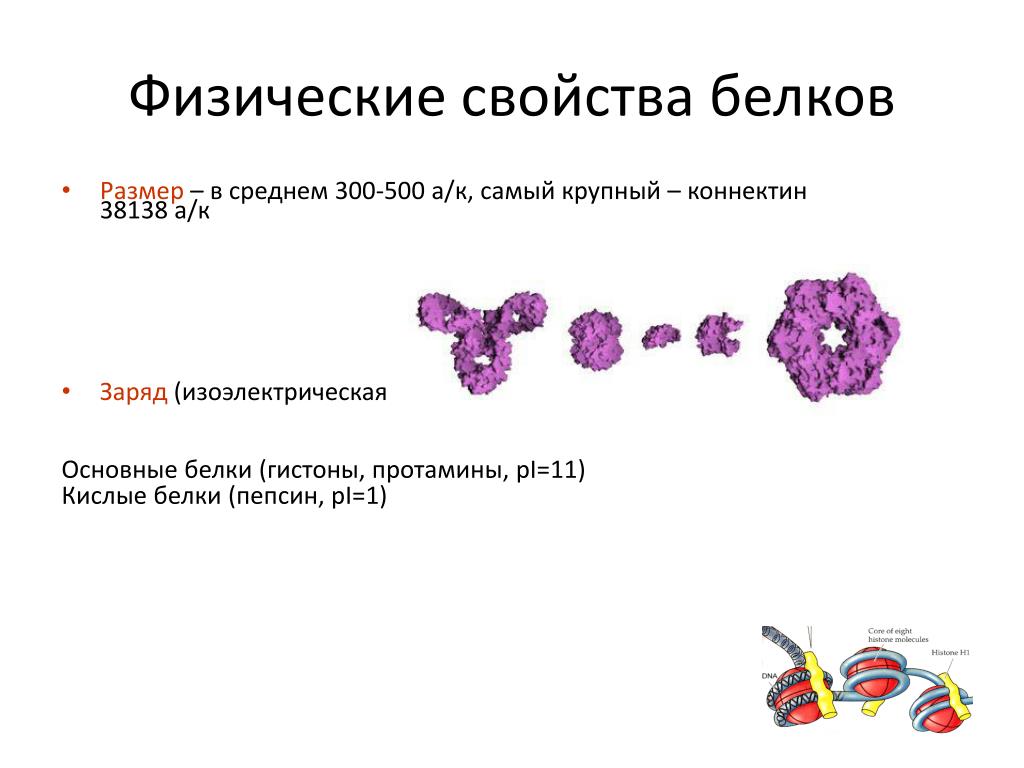
Все биохимические реакции в организмах протекают с участием ферментов. Ферменты — это белки-катализаторы. Значит, белки в живых организмах выполняют каталитическую функцию. Примерами таких катализаторов могут служить пищеварительные ферменты, участвующие в переваривании пищи: пепсин, липаза, амилаза, мальтаза.
Важнейшая функция белков — защитная. Особые белки — антитела и антитоксины — участвуют в формировании иммунитета. Антитела обезвреживают проникшие в организм бактерии, а антитоксины нейтрализуют их яды.
Белок гемоглобин выполняет транспортную функцию. Он переносит кислород от органов дыхания к тканям.
Двигательная функция некоторых белков обеспечивает сокращение мышц и все движения живых организмов.
При нехватке пищи белки могут выполнять энергетическую функцию. При расщеплении \(1\) г белка выделяется \(17,6\) кДж энергии.
Белки выполняют сигнальную, рецепторную, регуляторную и другие функции.
Источники:
Рис. 1. Денатурация белка https://image.shutterstock.com/image-illustration/structure-normal-disassembled-protein-600w-1507923140.jpg
Рис. 2. Биуретовая реакция
Рис. 3. Ксантапротеиновая реакция
Свойства белка открывают безграничный простор для творчества — Российская газета
Опытные кулинары хвалятся, что могут целый год готовить завтраки из яиц и ни разу не повториться. Я кулинар не настолько опытный, опять же и семья не поймёт, если в число блюд из яиц не попадут оладьи. Но согласна, что такое возможно. Биохимические и физические свойства яйца дают безграничный простор для творчества.
Шокирующий факт: наш вид массово употребляет в пищу женские половые клетки другого вида. Никакого преувеличения, для биолога яйцо птицы — это гигантская яйцеклетка в спецупаковке, созревающая вне тела матери. Потому оно так и питательно, что будущие петухи и курицы ни в чём не должны терпеть недостатка, пока не вылупятся.
В первую очередь яйцо — это, конечно, белок. Собственно, белки как класс соединений и названы были в честь белой части яйца. Белок яйца защищает эмбрион от повреждений и бактериальных инфекций, а заодно служит резервным запасом питательных веществ. Больше всего в нём белка под названием овальбумин, есть ещё овотрансферрин и лизоцим — они обладают антибактериальными свойствами, а овотрансферрин вдобавок снабжает эмбрион ионами железа. Яйцо на сковородке не растекается тонким слоем, как вода, благодаря гликопротеину овомуцину — в сыром белке его 2-3 %, но именно овомуцин делает его гелеобразным. Гликопротеин с похожим названием овомукоид — главный аллерген куриных яиц; аллергия бывает и на предыдущие три белка, а также на компоненты желтка, но овомукоид — любимая мишень иммунной системы. Поскольку белок вязкий, его можно взбивать в крепкую пену. Если с сахаром, получится безе, если подсолить — компонент омлета.
В 1800 году химик и министр просвещения наполеоновской Франции Антуан де Фуркруа описал особенные вещества, которые под действием кислот или высоких температур характерным образом свёртываются (по-научному — коагулируют) и содержатся, например, в яйцах или человеческой крови. Де Фуркруа назвал их альбуминоидами — от латинского albus ovi, буквально «яичный белок». Отсюда пошло и русское «белок», и немецкое Eiweiss, а международное protein предложил Йенс Берцелиус лишь в 1838 году.
Де Фуркруа назвал их альбуминоидами — от латинского albus ovi, буквально «яичный белок». Отсюда пошло и русское «белок», и немецкое Eiweiss, а международное protein предложил Йенс Берцелиус лишь в 1838 году.
Желток — основной пищевой запас для эмбриона птички. Белки в нём тоже есть, но ещё больше жиров. Жёлтым его делают лютеин и зеа — ксантин, относящиеся к каротиноидам (то есть родичам витамина А). Да, в желтке есть холестерин, но кроме него — много витаминов: А, D, Е, К, витамины группы В. Так что все эти натуральные маски для волос и кожи на сырых желтках — дело хорошее, да и есть яйца скорее полезно, чем вредно, если соблюдать меру.
Некоторых моих родственниц беспокоят яйца с необыкновенно яркими, почти оранжевыми желтками: «Курам что-то добавляют в корм!» Добавляют, как правило, натуральные экстракты каротиноидов, например, из лепестков календулы. Производители этого и не скрывают, даже наоборот — подчёркивают: каротиноиды полезны. Если давать курам экстракты красящих веществ перца, можно получить яйца с красными желтками (хотя не думаю, что это хорошая бизнес-идея). Курица на вольном выгуле несёт яйца, цвет желтка которых варьируется очень сильно в зависимости от того, что она клюёт.
Курица на вольном выгуле несёт яйца, цвет желтка которых варьируется очень сильно в зависимости от того, что она клюёт.
Белок яйца дал название белкам, а желток («лекитос» по-гречески) — лецитинам. Лецитины, как известно всем, кто читал составы пищевых продуктов на упаковках, — эмульгаторы: один конец их молекулы полярный, то есть охотно контактирует с водой, а другой неполярный, «жирный». Когда надо получить эмульсию жира в воде, лецитины незаменимы. В желтках их много, и это одна из причин, по которой их добавляют в соусы — тот же майонез.
Специфический «яичный» запах формирует то же вещество, что и запах тухлых яиц, — сероводород. В белках всегда есть сульфидные группы, и чем больше сульфида высвобождается при варке или чересчур длительном хранении, тем сильнее аромат. Кстати, сероватый налёт на поверхности желтка в яйце, которое слишком долго варили, — это сульфид железа. Он образуется из железа, содержащегося в желтке, и сероводорода белка. Некрасиво, но для здоровья не вредно.
Скорлупа состоит из кристаллов карбоната кальция, погружённых в белковую матрицу. Мельчайшие поры в ней позволяют яйцу «дышать». Интересный нюанс: сваренное яйцо, которое до этого хранилось долго, чистится легче, чем яйцо из свежих запасов, потому что при хранении из него улетучивается углекислый газ и внутренняя среда становится более щелочной. А в щелочной среде белки слабее взаимодействуют со скорлупой. Коричневатая скорлупа окрашена пигментом протопорфирином.
На яйцах из магазина бывают буквы и цифры, например С3 или Д1. Д — «диетическое», не старше 7 дней. С — «столовое», для них срок реализации 25 дней. Дальше идёт категория по массе: В (высшая), О (отборное яйцо), первая, вторая, третья. Яйцо высшей категории весит не меньше 75 граммов, третьей — от 35 до 44 граммов. Разброс немаленький, поэтому на кулинарных форумах часто указывают, яйца какой категории пошли в тесто для данного пирога.
Что происходит при термообработке яйца, будь то варка в скорлупе, поджаривание яичницы или запекание омлета в духовке? С этим никаких проблем даже у школьников: классический пример денатурации белков — то самое, что наблюдал де Фуркруа. Аккуратные шарики-глобулы раскручиваются, нити перепутываются и образуют тугую сеть (сильно не пережариваем, а то яичница будет как подошва). Кстати, денатурированный яичный белок усваивается гораздо лучше сырого.
Аккуратные шарики-глобулы раскручиваются, нити перепутываются и образуют тугую сеть (сильно не пережариваем, а то яичница будет как подошва). Кстати, денатурированный яичный белок усваивается гораздо лучше сырого.
Умение жарить яичницу — главный тест на личную независимость, а сама яичница — лидер сразу в двух номинациях: на звание Самого Любимого Завтрака и Самого Осточертевшего. Поэтому вооружаемся приобретённым знанием и идём изучать рецепты блюд из яиц. Пусть не на каждый день года, но с полдюжины знать надо.
Омлет «Пуляр»
Берём четыре яйца, отделяем белки от желтков. Желтки слегка взбиваем, добавляем две ложки молока, соль и перец по вкусу. Белки тоже подсаливаем и взбиваем в крепкую пену (при вытаскивании венчика на поверхности должен получиться острый пик). Выливаем желтки на большую сковородку, разогретую и слегка смазанную маслом. Дожидаемся, пока они запекутся, уменьшаем огонь до минимума и выкладываем белки. Крышкой не накрываем и ждём 8-10 минут (белки должны слегка схватиться — так, чтобы не прилипали к пальцу). Разрезаем омлет пополам и складываем белком внутрь.
Разрезаем омлет пополам и складываем белком внутрь.
Ученые объяснили противораковые свойства популярного напитка
https://ria.ru/20210212/chay-1597213878.html
Ученые объяснили противораковые свойства популярного напитка
Ученые объяснили противораковые свойства популярного напитка — РИА Новости, 13.04.2021
Ученые объяснили противораковые свойства популярного напитка
Биологи выяснили, что соединение, содержащееся в зеленом чае, стабилизирует противораковый белок — «хранитель генома», который подавляет рост раковых клеток… РИА Новости, 13.04.2021
2021-02-12T14:26
2021-02-12T14:26
2021-04-13T18:51
наука
зеленый чай
биология
рак
здоровье
китай
сша
/html/head/meta[@name=’og:title’]/@content
/html/head/meta[@name=’og:description’]/@content
https://cdn21.img.ria.ru/images/07e5/02/0c/1597210819_0:116:1440:926_1920x0_80_0_0_b263db72ddd33a4dbd43ee2a624d5442.jpg
МОСКВА, 12 фев — РИА Новости. Биологи выяснили, что соединение, содержащееся в зеленом чае, стабилизирует противораковый белок — «хранитель генома», который подавляет рост раковых клеток. Результаты исследования опубликованы в журнале Nature Communications.В зеленом чае в больших количествах содержится вещество, которое устраняет ущерб, причиненный метаболизмом кислорода, — природный антиоксидант галлат эпигаллокатехина (EGCG).Исследователи из США и Китая установили, что EGCG способен напрямую взаимодействовать с противораковым белком p53, повышая его уровень в организме. Белок p53 часто называют хранителем генома за его способность восстанавливать повреждения ДНК и разрушать раковые клетки.»Как молекулы p53, так и EGCG чрезвычайно интересны. Более чем в половине случаев рака человека у пациентов обнаруживают мутации в p53, а EGCG является основным антиоксидантом в зеленом чае, популярном во всем мире напитке, — приводятся в пресс-релизе Политехнического института Ренсселера слова руководителя исследования профессора Ван Чунью (Chunyu Wang).
Биологи выяснили, что соединение, содержащееся в зеленом чае, стабилизирует противораковый белок — «хранитель генома», который подавляет рост раковых клеток. Результаты исследования опубликованы в журнале Nature Communications.В зеленом чае в больших количествах содержится вещество, которое устраняет ущерб, причиненный метаболизмом кислорода, — природный антиоксидант галлат эпигаллокатехина (EGCG).Исследователи из США и Китая установили, что EGCG способен напрямую взаимодействовать с противораковым белком p53, повышая его уровень в организме. Белок p53 часто называют хранителем генома за его способность восстанавливать повреждения ДНК и разрушать раковые клетки.»Как молекулы p53, так и EGCG чрезвычайно интересны. Более чем в половине случаев рака человека у пациентов обнаруживают мутации в p53, а EGCG является основным антиоксидантом в зеленом чае, популярном во всем мире напитке, — приводятся в пресс-релизе Политехнического института Ренсселера слова руководителя исследования профессора Ван Чунью (Chunyu Wang). — Теперь мы обнаружили ранее неизвестное прямое взаимодействие между ними. Наша работа помогает объяснить, как EGCG усиливает противораковую активность p53, и открывает двери к разработке противораковых лекарств с EGCG-подобными соединениями».Ван Чунью, сотрудник Центра биотехнологии и междисциплинарных исследований института Ренсселера, давно исследует р53 с помощью метода спектроскопии ядерного магнитного резонанса для изучения конкретных механизмов болезни Альцгеймера и рака. Он называет его «возможно, самым важным белком для борьбы с раком».Белок р53 выполняет несколько хорошо известных противораковых функций, включая остановку роста раковых клеток, активацию восстановления ДНК и запуск запрограммированной гибели клеток (апоптоза), если повреждение ДНК не может быть устранено.Один конец белка, известный как N-концевой домен, имеет гибкую форму и потенциально может выполнять несколько функций в зависимости от его взаимодействия с другими молекулами. Обычно после образования в организме р53 быстро разлагается, когда N-концевой домен соединяется с белком MDM2.
— Теперь мы обнаружили ранее неизвестное прямое взаимодействие между ними. Наша работа помогает объяснить, как EGCG усиливает противораковую активность p53, и открывает двери к разработке противораковых лекарств с EGCG-подобными соединениями».Ван Чунью, сотрудник Центра биотехнологии и междисциплинарных исследований института Ренсселера, давно исследует р53 с помощью метода спектроскопии ядерного магнитного резонанса для изучения конкретных механизмов болезни Альцгеймера и рака. Он называет его «возможно, самым важным белком для борьбы с раком».Белок р53 выполняет несколько хорошо известных противораковых функций, включая остановку роста раковых клеток, активацию восстановления ДНК и запуск запрограммированной гибели клеток (апоптоза), если повреждение ДНК не может быть устранено.Один конец белка, известный как N-концевой домен, имеет гибкую форму и потенциально может выполнять несколько функций в зависимости от его взаимодействия с другими молекулами. Обычно после образования в организме р53 быстро разлагается, когда N-концевой домен соединяется с белком MDM2.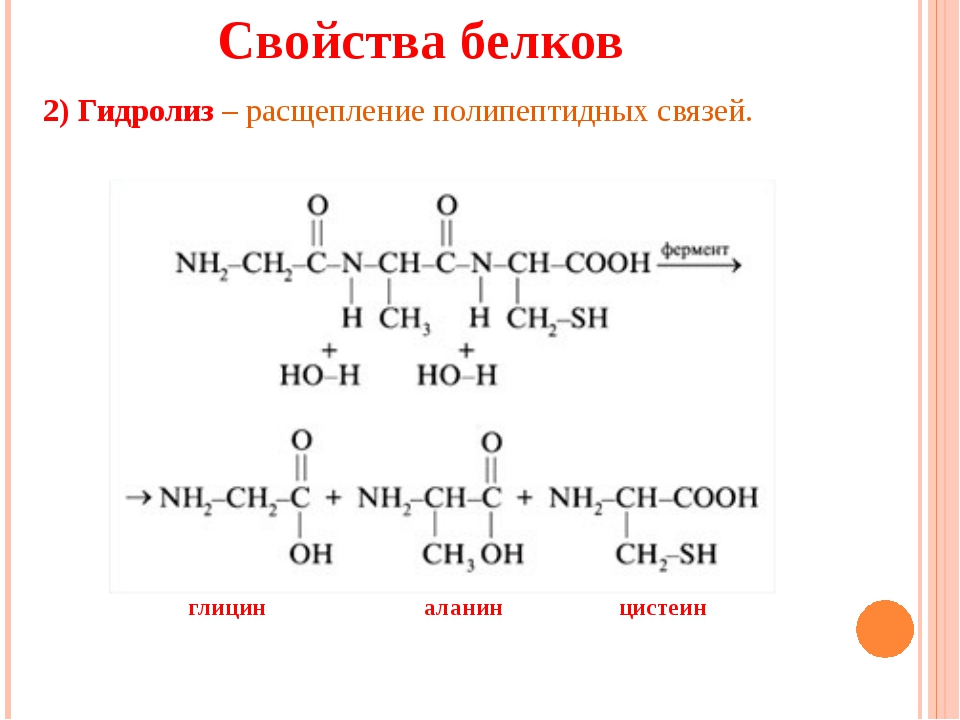 Этот регулярный цикл производства и деградации удерживает уровни p53 на достаточно низком уровне. Ученые обнаружили, что взаимодействие между EGCG и p53 предохраняет белок от деградации.»И EGCG, и MDM2 связываются в одном и том же месте — на N-концевом домене p53, поэтому EGCG конкурирует с MDM2, — объясняет ученый. — Когда EGCG связывается с p53, белок не разлагается через взаимодействие с MDM2, поэтому при прямом взаимодействии с EGCG уровень p53 будет увеличиваться, а это означает, что его противораковая функция будет высокой. Это очень важное взаимодействие».Авторы отмечают, что им удалось впервые объяснить молекулярный механизм антиракового эффекта зеленого чая, давно доказанного в обсервационных исследованиях.
Этот регулярный цикл производства и деградации удерживает уровни p53 на достаточно низком уровне. Ученые обнаружили, что взаимодействие между EGCG и p53 предохраняет белок от деградации.»И EGCG, и MDM2 связываются в одном и том же месте — на N-концевом домене p53, поэтому EGCG конкурирует с MDM2, — объясняет ученый. — Когда EGCG связывается с p53, белок не разлагается через взаимодействие с MDM2, поэтому при прямом взаимодействии с EGCG уровень p53 будет увеличиваться, а это означает, что его противораковая функция будет высокой. Это очень важное взаимодействие».Авторы отмечают, что им удалось впервые объяснить молекулярный механизм антиракового эффекта зеленого чая, давно доказанного в обсервационных исследованиях.
https://ria.ru/20200109/1563213655.html
https://ria.ru/20190924/1559029532.html
китай
сша
РИА Новости
7 495 645-6601
ФГУП МИА «Россия сегодня»
https://xn--c1acbl2abdlkab1og.xn--p1ai/awards/
2021
РИА Новости
internet-group@rian. ru
ru
7 495 645-6601
ФГУП МИА «Россия сегодня»
https://xn--c1acbl2abdlkab1og.xn--p1ai/awards/
Новости
ru-RU
https://ria.ru/docs/about/copyright.html
https://xn--c1acbl2abdlkab1og.xn--p1ai/
РИА Новости
7 495 645-6601
ФГУП МИА «Россия сегодня»
https://xn--c1acbl2abdlkab1og.xn--p1ai/awards/
https://cdn25.img.ria.ru/images/07e5/02/0c/1597210819_132:0:1412:960_1920x0_80_0_0_ca9b08982951823b43fd872e6b878fd5.jpg
РИА Новости
7 495 645-6601
ФГУП МИА «Россия сегодня»
https://xn--c1acbl2abdlkab1og.xn--p1ai/awards/
РИА Новости
7 495 645-6601
ФГУП МИА «Россия сегодня»
https://xn--c1acbl2abdlkab1og.xn--p1ai/awards/
зеленый чай, биология, рак, здоровье, китай, сша
МОСКВА, 12 фев — РИА Новости. Биологи выяснили, что соединение, содержащееся в зеленом чае, стабилизирует противораковый белок — «хранитель генома», который подавляет рост раковых клеток. Результаты исследования опубликованы в журнале Nature Communications.
Результаты исследования опубликованы в журнале Nature Communications.
В зеленом чае в больших количествах содержится вещество, которое устраняет ущерб, причиненный метаболизмом кислорода, — природный антиоксидант галлат эпигаллокатехина (EGCG).
Исследователи из США и Китая установили, что EGCG способен напрямую взаимодействовать с противораковым белком p53, повышая его уровень в организме. Белок p53 часто называют хранителем генома за его способность восстанавливать повреждения ДНК и разрушать раковые клетки.
«Как молекулы p53, так и EGCG чрезвычайно интересны. Более чем в половине случаев рака человека у пациентов обнаруживают мутации в p53, а EGCG является основным антиоксидантом в зеленом чае, популярном во всем мире напитке, — приводятся в пресс-релизе Политехнического института Ренсселера слова руководителя исследования профессора Ван Чунью (Chunyu Wang). — Теперь мы обнаружили ранее неизвестное прямое взаимодействие между ними. Наша работа помогает объяснить, как EGCG усиливает противораковую активность p53, и открывает двери к разработке противораковых лекарств с EGCG-подобными соединениями».
9 января 2020, 13:54НаукаУченые выяснили, почему любители чая живут дольше
Ван Чунью, сотрудник Центра биотехнологии и междисциплинарных исследований института Ренсселера, давно исследует р53 с помощью метода спектроскопии ядерного магнитного резонанса для изучения конкретных механизмов болезни Альцгеймера и рака. Он называет его «возможно, самым важным белком для борьбы с раком».
Белок р53 выполняет несколько хорошо известных противораковых функций, включая остановку роста раковых клеток, активацию восстановления ДНК и запуск запрограммированной гибели клеток (апоптоза), если повреждение ДНК не может быть устранено.
Один конец белка, известный как N-концевой домен, имеет гибкую форму и потенциально может выполнять несколько функций в зависимости от его взаимодействия с другими молекулами. Обычно после образования в организме р53 быстро разлагается, когда N-концевой домен соединяется с белком MDM2. Этот регулярный цикл производства и деградации удерживает уровни p53 на достаточно низком уровне. Ученые обнаружили, что взаимодействие между EGCG и p53 предохраняет белок от деградации.
Ученые обнаружили, что взаимодействие между EGCG и p53 предохраняет белок от деградации.
«И EGCG, и MDM2 связываются в одном и том же месте — на N-концевом домене p53, поэтому EGCG конкурирует с MDM2, — объясняет ученый. — Когда EGCG связывается с p53, белок не разлагается через взаимодействие с MDM2, поэтому при прямом взаимодействии с EGCG уровень p53 будет увеличиваться, а это означает, что его противораковая функция будет высокой. Это очень важное взаимодействие».
Авторы отмечают, что им удалось впервые объяснить молекулярный механизм антиракового эффекта зеленого чая, давно доказанного в обсервационных исследованиях.
24 сентября 2019, 09:51НаукаУченые обнаружили неожиданное свойство зеленого чая
Ученые создали белки, свойства которых можно изменять светом — Газета.Ru
Исследователи разработали флуоресцентные белки, свойствами которых можно управлять с помощью оранжевого и зеленого света. Эти белки помогут ученым исследовать процессы жизнедеятельности в живых клетках. Работа проходила в рамках проекта, который поддерживается грантом Российского научного фонда (РНФ), а ее результаты были опубликованы в журнале Nature Methods.
Работа проходила в рамках проекта, который поддерживается грантом Российского научного фонда (РНФ), а ее результаты были опубликованы в журнале Nature Methods.
Флуоресцентные белки – это белки, которые интенсивно светятся в видимом диапазоне спектра, где длина волн составляет от 390 до 700 нанометров. Природные функции таких белков достаточно разнообразны, например, некоторые виды медуз с помощью зеленых флуоресцентных пятнышек приманивают к себе еду – различные мелкие организмы. Оптическими свойствами некоторых флуоресцентных белков – их называют фотопереключаемыми – можно управлять с помощью света. Например, эти белки можно «включать» и «выключать», что широко используется в так называемой флуоресцентной микроскопии сверхвысокого разрешения (наноскопии) – новой группе методов, позволяющих получать чрезвычайно детальные изображения внутриклеточных структур. Обычно для такой микроскопии ученые используют синее и фиолетовое облучение, которое очень токсично для клеток: оно нарушает их нормальную физиологию и даже вызывает гибель.
«Мы впервые создали фотопереключаемые флуоресцентные белки, оптические свойства которых можно контролировать зеленым и оранжевым, а не сине-фиолетовым светом. Его преимущество состоит в том, что он почти не причиняет вреда клеткам. Мы использовали новые белки для наблюдения динамики цитоскелета в живых клетках», – рассказал один из авторов статьи Александр Мишин, руководитель проекта РНФ, кандидат биологических наук, старший научный сотрудник Института биоорганической химии имени академиков М.М. Шемякина и Ю.А. Овчинникова РАН.
Для создания флуоресцентных белков ученые изменяли их (направленный и случайный мутагенез) с помощью такого метода, как полимеразная цепная реакция, которая позволяет добиться значительного увеличения малых концентраций определенных фрагментов ДНК. Также ученые клонировали белки, после чего под микроскопом отбирали из полученных бактериальных колоний самые удачные. Авторы проанализировали результаты уже проведенных другими биологами экспериментов и выяснили, как приблизительно нужно изменить микроокружение хромофора (остатка ароматической аминокислоты, который отвечает за поглощение света в белке), чтобы заставить его проявить способность к фотопереключению.
Помимо ожидаемого эффекта, однако, есть и побочные, например, снижается яркость белка. Тогда случайный мутагенез позволяет найти дополнительные мутации, компенсирующие побочные эффекты при сохранении целевого.
Разработанные белки называются репортерными, так как выполняют роль «шпионов» в клетках. Их присоединяют к другим белкам и таким образом следят за ними в живой клетке. Полученная детальная информация может быть использована как в фундаментальных, так и в биомедицинских исследованиях. Например, у больного раком пациента опухолевые клетки демонстрируют сильные нарушения клеточной подвижности и динамических перестроек цитоскелета – каркаса, находящегося в цитоплазме живой клетки. При этом изучение этих процессов со сверхвысоким разрешением (наноскопия) затруднено в живых клетках из-за слишком интенсивного облучения образца, поэтому для таких целей нужно использовать методы с меньшей токсичностью для организма.
Авторы использовали свою разработку для микроскопии сверхвысокого разрешения RESOLFT. У полученных белков есть особенность: у них очень эффективное фотопереключение, то есть белки «включаются» и «выключаются» за миллисекунды. Такое свойство подходит не всем методам микроскопии, в некоторых такая скорость будет только мешать. В RESOLFT цикл включения-выключения повторяется многократно для соседних точек, сканируемых лазерными пучками. Чем лучше флуоресцентная метка переключается, тем быстрее удается снять полное изображение, так как на фотопереключение в каждой точке нужно меньше времени.
У полученных белков есть особенность: у них очень эффективное фотопереключение, то есть белки «включаются» и «выключаются» за миллисекунды. Такое свойство подходит не всем методам микроскопии, в некоторых такая скорость будет только мешать. В RESOLFT цикл включения-выключения повторяется многократно для соседних точек, сканируемых лазерными пучками. Чем лучше флуоресцентная метка переключается, тем быстрее удается снять полное изображение, так как на фотопереключение в каждой точке нужно меньше времени.
«Созданные нами флуоресцентные белки позволяют проводить сверхразрешающую микроскопию без вреда для живой клетки, что открывает возможность исследования динамических процессов в ней», – заключил ученый.
Работа проходила в сотрудничестве с учеными из Королевского технологического института (Швеция) и Медицинского колледжа Альберта Эйнштейна (США).
Свойства и цветовая палитра флуоресцентных белков
Введение во флуоресцентные белки
Открытие в начале 1960-х зелёного флуоресцентного белка провозгласило новую эру в клеточной биологии, поскольку позволило исследователям применять методы молекулярного клонирования, присоединяя флуророфорный компонент к многочисленным белкам и энзимам, чтобы наблюдать с помощью оптических микроскопов процессы в живых клетках. В сочетании с последними техническими достижениями широкопольной флуоресцентной и конфокальной микроскопии, включая быстродействующие цифровые камеры, работающие в условиях низкой освещённости, и лазерные системы с многоканальным слежением, зелёный флуоресцентный белок и его смещённые по цвету генетические дериваты оказались настолько эффективными в многочисленных экспериментах по наблюдению живых клеток, что их значение трудно переоценить.
Рис. 1. Мультиокрашивание живых клеток флуоресцентными белками
Работая во Фрайдей Харбор Лабораториз (Friday Harbor Laboratories) университета Вашингтона, Осаму Шимомура (Osamu Shimomura) и Фрэнк Джонсон (Frank Johnson) выделили в 1961 году кальций-зависимый биолюминесцентный белок медузы Aequorea victoria, назанный ими экварином (aequorin). Во время процедуры выделения наблюдался второй белок, который не имел синего биолюминесцентного свечения экварина, но при облучении ультрафиолетом флуоресцировал зелёным. Благодаря этому свойству белок был неофициально назван зелёным флуоресцентным белком (GFP). В течение двух последующих десятилетий было установлено, что экварин и зелёный флуоресцентный белок функционируют вместе в органах свечения медуз, преобразовывая индуцируемые кальцием люминесцентные сигналы в зелёную флуоресценцию, характерную для этого вида.
Хотя ген для зелёного флуоресцентного белка был впервые клонирован в 1992 году, его потенциал, как молекулярного зонда, оценили лишь несколько лет спустя, когда продукты слияния стали применяться для генной экспрессии в бактериях и нематодах. С тех пор зелёный флуоресцентный белок стал синтезироваться для производства огромного количества различных окрашенных мутантов, гибридных белков и биосенсоров, обобщённо называемых флуоресцентными белками. Позже флуоресцентные белки были выделены и из других видов, что способствовало дальнейшему расширению цветовой палитры. С развитием технологии флуоресцентных белков значение этого генетически кодированного флуророфора для широкого круга приложений, помимо простого наблюдения меченных биомолекул в живых клетках, в полной мере стало осознаваться только сейчас.
На рисунке 1 приведены два примера мультиокрашивания живых клеток флуоресцентными белками, где слияние происходит на субклеточном уровне (на уровне органелл). Эпителиальная клетка кортикального проксимального канальца почки опоссума (ОК линия), представленная на рисунке 1(а), была трансфектирована набором флуоресцентных белков, соединённых с пептидными сигналами, осуществляющими опосредованный перенос белков либо к ядрам (усиленный (т. е. с усиленным свечением) голубой флуоресцентный белок, ECFP), либо к митохондриям (флуоресцентный белок DsRed: DsRed2FP), либо к сети микроканальцев (усиленный зелёный флуоресцентный белок, EGFP). Аналогичный образец, состоящий из клеток цервикальной аденокарциномы человека (HeLa линия), представлен на рисунке 1(b). Клетки HeLa были трансфектированы векторами, несущими кодирующие последовательности усиленного голубого и жёлтого (EYFP) флуоресцентных белков для соединеня с комплексом Гольджи и ядром, соответственно, а также вариант флуоресцентного белка морской анемоны Discosoma striata, DsRed2FP, нацеленный на митохондриальную сеть.
Зелёный флуоресцентный белок и его мутировавшие аллельные формы (синий, голубой и жёлтый флуоресцентный белок) используются для создания флуоресцентных химерных белков, которые, после трансфекции созданными векторами, могут быть экспрессированы в живых клетках, тканях и целых организмах. Красные флуоресцентные белки, выделенные из других видов, включая организмы коралловых рифов, имеют такую же ценность. Методика с применением флуоресцентных белков свободна от таких проблем, как очистка, мечение и введение меченых белков в клетки, а также от разработки специальных антигенов для поверхностных и внутренних антител.
Свойства и модификации зелёного флуоресцентного белка медузы Aequorea victoria
Существенным аспектом в свечении зелёного флуоресцентного белка является тот факт, что для развития и поддержания флуоресценции необходима целая нативная пептидная структура весом в 27 килодальтонов. Примечательно, что сам флуророфор образуется из триплета соседних аминокислот: остатков серина, тирозина и глицина в позициях 65, 66 и 67 (обозначаемых Сер-65 (Ser65), Тир-66 (Tyr66) и Гли-67 (Gly67), см. рисунок 2) Хотя этот несложный аминокислотный рисунок широко распространён в природе, он не всегда вызывает флуоресценцию. Уникальность флуоресцентного белка заключается в том, что пептидный триплет находится в центре удивительно стабильной бочкообразной структуры, состоящей из 11 бета-слоёв, свёрнутых в цилиндр.
В гидрофобном окружении в центре зелёного флуоресцентного белка происходит реакция между карбоксильным углеродом в Сер-65 и азотом аминогруппы в Гли-67, которая приводит к образованию имидазолин-5-онового гетероциклического азотного кольца (как показано на рисунке 2). Дальнейшее окисление ведёт к объединению имидазолинового кольца с Тир-66 и созреванию флуоресцентного вида. Важно отметить, что нативный флуророфор зелёного флуоресцентного белка существует в двух состояниях. Преобладающая протонированная форма имеет максимум возбуждения на 395 нанометрах, а менее распространенная непротонированная форма поглощает приблизительно на 475 нанометрах. Тем не менее, независимо от длины волны возбуждения пик флуоресцентного свечения, хотя размытый и широкий, приходится на 507 нанометров.
Рис. 2. Образование имидазолин-5-онового гетероциклического азотного кольца
Два основных свойства флуророфора флуоресцентного белка являются определяющими для его применения в качестве зонда. Первое — это сложность фотофизических свойств зелёного флуоресцентного белка, обеспечивающая существование значительного числа модификаций этой молекулы. Многие исследования были сосредоточены на тонкой настройке флуоресценции нативного зелёного флуоресцентного белка для получения многочисленных молекулярных красителей, но трудно переоценить применение этого белка в качестве исходного материала для создания новых флуорофоров с улучшенными свойствами, что, конечно, имеет огромное значение и потенциал. Второе свойство — сильная зависимость флуоресценции зелёного флуоресцентного белка от молекулярной структуры, окружающей трипептидный флуророфор.
Денатурация зелёного флуоресцентного белка разрушает флуоресценцию, что вполне предсказуемо, а мутации остатков, окружающих трипептидный флуорофор, могут значительно изменить его флуоресцентные свойства. Удивительная стабильность бета-бочонка, в котором находятся аминокислотные остатки, приводит к очень высокому квантовому выходу флуоресценции (до 80 процентов). Эта очень плотная белковая структура обеспечивает неизменность флуоресцентных свойств даже при изменениях pH, температуры и в присутствии денатурирующих агентов, таких как мочевина. Но при мутациях зелёного флуоресцентного белка эта стабильность обычно падает, что сказывается на флуоресценции, приводя к уменьшению квантового выхода и большей зависимости от окружающих условий. Хотя в некоторой степени это преодолевается дополнительными мутациями, дериваты флуоресцентных белков обычно более чувствительны к окружающим условиям, чем нативные виды. Эти изменения должны приниматься во внимание при организации экспериментов с генетическими вариантами.
Чтобы приспособить флуоресцентные белки к наблюдению различных систем млекопитающих, были произведены некоторые модификации исходного зелёного флуоресцентного белка, которые сегодня встречаются в большинстве применяемых вариантов. Первым шагом была оптимизация созревания белка для флуоресценции при температуре 37 градусов по Цельсию. Созревание флуророфора исходного типа вполне эффективно при 28 градусах, но увеличение температуры до 37 градусов приводит к существенному ухудшению созревания и уменьшению флуоресценции. Мутация остатка фенилаланина в позиции 64 (Фен-64 (Phe64)) в лейцин улучшает созревание белка для флуоресценции при 37 градусах, которая, по меньшей мере, не хуже наблюдаемой при 28 градусах. Эта мутация присутствует в большинстве применяемых вариантов флуоресцентных белков, полученных из Aequorea victoria, но это, как выяснилось при открытии других вариантов, не единственная мутация, улучшающая укладку при 37 градусах.
Улучшение созревания при 37 градусах было дополнено оптимизацией использования кодона для экспрессии в клетках млекопитающих, что тоже повысило яркость зелёного флуоресцентного белка, экспрессированного в этих клетках. В общей сложности, для усиления экспрессии в тканях человека в кодирующую последовательность было введено более 190 молчащих мутаций. Вставкой валина в качестве второй аминокислоты был введён участок инициации трансляции Козака (содержащий нуклеотидную последовательность A/GCCAT). Наряду с другими, обсуждаемыми ниже, эти усовершенствования привели к созданию очень эффективных красителей для наблюдения живых клеток млекопитающих и являются общими для всех применяемых сегодня флуоресцентных зондов, полученных из исходного белка медузы.
Цветовая палитра флуоресцентных белков
Спектральные кривые флуоресцентного свечения многочисленных генетических вариантов флуоресцентных белков, которые удалось получить, перекрывают практически весь видимый диапазон (см. таблицу 1). Усилия по мутагенезу исходного зелёного флуоресцентного белка медузы Aequorea victoria привели к созданию флуоресцентных красителей в цветовой гамме от синего до жёлтого и к тому, что они стали самыми распространёнными в биологических исследованиях репортерными группами in vivo. Флуоресцентные белки бо?льших длин волн, светящиеся в оранжевом и красном диапазонах, были получены из морской анемоны, Discosoma striata, и рифовых кораллов, принадлежащих к классу Anthozoa (коралловых полипов). И, тем не менее, разработка аналогичных белков с голубым, зелёным, жёлтым, оранжевым и тёмно-красным флуоресцентным свечением не прекращается. Сейчас разработки ведутся в направлении повышения яркости и стабильности флуоресцентных белков, что, в итоге, способствует повышению их эффективности.
Зелёные флуоресцентные белки
Хотя исходный зелёный флуоресцентный белок флуоресцирует достаточно интенсивно и чрезвычайно стабилен, его максимум возбуждения лежит близко к ультрафиолету. Поскольку ультрафиолетовый свет требует специальных оптических условий и способен нанести вред живым клеткам, он часто бывает непригоден для наблюдения живых клеток в оптической микроскопии. К счастью, максимум возбуждения зелёного флуоресцентного белка можно сдвинуть к 488 нанометрам (в голубой диапазон) с помощью точечной мутации, заменяющей серин в позиции 65 на остаток треонина (S65T). Эта мутация присутствует в большинстве распространённых вариантов зелёного флуоресцентного белка, называемых усиленным GFP (EGFP), которые выпускаются серийно в широком диапазоне векторов, предлагаемых БД Биосайенсис Клонтек (BD Biosciences Clontech), одним из лидеров по производству флуоресцентных белков. Более того, усиленная версия может наблюдаться с помощью общедоступных наборов фильтров, разработанных для флюоресцеина, и является одной из наиболее ярких из доступных сегодня флуоресцентных белков. Благодаря этим свойствам усиленный зелёный флуоресцентный белок стал одним из наиболее распространённых красителей и не имеет себе равных в экспериментах с одной меткой. Единственным недостатком EGFP является слегка повышенная чувствительность к pH и некоторая тенденция к димеризации.
Помимо усиленного зелёного флуоресцентного белка при наблюдении живых клеток сегодня применяются и другие варианты. Одним из лучших с точки зрения фотостабильности и яркости, может быть, является белок Эмеральд (Emerald), но применение этого варианта ограничено тем, что он не выпускается серийно. Некоторые производители предлагают гуманизированные варианты зелёного флуоресцентного белка, которые имеют определённые преимущества в экспериментах по резонансному переносу энергии флуоресценции (FRET). В результате замены фенилаланинового остатка в позиции 64 на лейцин (F64L; GFP2) получается мутант, который сохраняет пик возбуждения на 400 нанометрах и является эффективным партнёром усиленному жёлтому флуоресцентному белку. Вариант с мутацией S65C (обычная замена цистеина на серин), имеющий пик возбуждения на 474 нанометрах, стал выпускаться серийно в качестве более эффективного FRET партнёра для усиленного синего флуоресцентного белка, чем усиленный зелёный вариант с красным смещением. Наконец, белок рифового коралла, называемый ZsGreen1 (ZsЗелёный1), с эмиссионным пиком на 505 нанометрах был разработан в качестве замены усиленному зелёному флуоресцентному белку. При экспрессировании в клетках млекопитающих свечение белка ZsЗелёный1 гораздо ярче EGFP, но он имеет ограниченное применение в получении слитых мутантов и, подобно остальным белкам рифовых кораллов, склонен к формированию тетрамеров.
Жёлтые флуоресцентные белки
Семейство жёлтых флуоресцентных белков было запущено после того, как обнаружилось, что в структуре зелёного флуоресцентного белка треониновый остаток 203 (Thr203) находится рядом с хромофором. Замена этого остатка на тирозин была введена для стабилизации дипольного момента возбуждённого состояния хромофора и привела к 20-нанометровому сдвигу в сторону длинных волн как спектра возбуждения, так и эмиссионного спектра. Дальнейшие усовершенствования привели к разработке усиленного жёлтого флуоресцентного белка (EYFP), который является одним из наиболее ярких и распространённых среди флуоресцентных белков. Благодаря своей яркости в сочетании со спектром свечения усиленный жёлтый флуоресцентный белок является превосходным кандидатом на применение в экспериментах по мультиокрашиванию во флуоресцентной микроскопии. Будучи спаренным с усиленным голубым флуоресцентным белком (ECFP) или GFP2, усиленный жёлтый флуоресцентный белок также показал свою эффективность в экспериментах по переносу энергии. Однако, жёлтый флуоресцентный белок слишком чувствителен к кислым средам и теряет приблизительно половину своего свечения при pH 6,5. К тому же, EYFP оказался чувствителен к ионам хлорида и фотообесцвечивается гораздо быстрее зелёных флуоресцентных белков.
Рис. 3. Спектральные кривые распространённых флуоресцентных белков
Дальнейшее усовершенствование архитектуры флуоресцентных белков для жёлтого свечения привело к решению некоторых проблем жёлтых красителей. Цитрин (Citrine), вариант жёлтого флуоресцентного белка, получился гораздо ярче EYFP и значительно устойчивее к фотообесцвечиванию, кислой среде и другим внешним воздействиям. Другой дериват с названием Венера (Venus) имеет самое быстрое созревание и на сегодня является одним из самых ярких вариантов жёлтого белка. Белок рифового коралла ZsYellow1 (ZsЖёлтый1), первоначально клонированный из Zoanthus, обитающих в Индийском и Тихом океанах, производит реалистичное жёлтое свечение и идеален для мультиокрашенных приложений. Подобно белку ZsЗелёный1, этот дериват не столь эффективен для образования соединений, как EYFP, и склонен к формированию тетрамеров. Многие из более стабильных вариантов жёлтого флуоресцентного белка оказались полезными для количественной оценки результатов FRET исследований и могут быть полезными и в других приложениях
На рисунке 3 представлены спектральные профили поглощения и испускания для многих часто встречаемых и серийно выпускаемых флуоресцентных белков, охватывающие диапазон от голубого до дальнего красного. Варианты, полученные из медузы Aequorea victoria, включая усиленные голубой, зелёный и жёлтый флуоресцентные белки, демонстрируют пик свечения на длинах от 425 до 525 нанометрах. Флуоресцентные белки, полученные из рифовых кораллов, DsRed2 (DsКрасный2) и HcRed1 (HcКрасный1) (обсуждаются ниже), испускают на более длинных волнах, но подвержены олигомеризационным артефактам в клетках млекопитающих.
Синие и голубые флуоресцентные белки
Синий и голубой варианты зелёного флуоресцентного белка получаются прямой модификацией тирозинового остатка в позиции 66 (Тир-66) в исходном флуророфоре (см. рисунок 2). Конверсия этой аминокислоты в гистидин приводит к синему свечению с максимумом на 450 нанометрах, а конверсия в триптамин — к основному пику флуоресценции на 480 нанометрах с плечом на длине около 500 нанометров. Оба красителя флуоресцируют весьма слабо, и для повышения эффективности укладки и общей яркости требуются вторичные мутации. Но даже с этими модификациями усиленные версии этого класса флуоресцентных белков (EBFP и ECFP) флуоресцируют лишь с 25-40-процентной яркостью в сравнении с усиленным зелёным флуоресцентным белком. К тому же, возбуждение синего и голубого флуоресцентного белка наиболее эффективно в нечасто используемом спектральном диапазоне, поэтому для них требуются специальные наборы фильтров и лазерные излучатели.
Несмотря на недостатки синего и голубого флуоресцентного белка широкий интерес к экспериментам по мультиокрашиванию и FRET привёл к росту их применения во многих приложениях. Это особенно касается усиленного голубого флуоресцентного белка, который может быть возбуждён вне своего пика аргоновым ионным лазером (на спектральной линии 457 нанометров) и гораздо более устойчив к фотообесцвечиванию, чем синий дериват. В отличие от других флуоресцентных белков, среди исследователей не было особенного интереса к созданию лучших красителей в синем диапазоне, и главные их усилия были направлены на разработку усовершенствованных голубых вариантов.
Рис. 4. Спектральные кривые спаренных для FRET флуоресцентных белков
Среди усовершенствованных голубых флуоресцентных белков AmCyan1 (AmГолубой1) и усиленный голубой вариант, называемый Cerulean (Лазурный) оказались наиболее перспективными. Полученный из рифового коралла, Anemonia majano, вариант флуоресцентного белка AmГолубой1 был оптимизирован кодонами человека для достижения относительно высокого уровня яркости и стойкости к фотообесцвечиванию при экспрессии в клетках млекопитающих по сравнению с усиленным голубым флуоресцентным белком. Но с другой стороны, этот краситель, подобно другим белкам рифовых кораллов, имеет тенденцию к образованию тетрамеров. Флуоресцентный краситель Лазурный был получен сайт-направленным мутагенезом усиленного голубого флуоресцентного белка для достижения более высокого коэффициента поглощения и квантового выхода. Лазурный минимум в 2 раза ярче усиленного голубого флуоресцентного белка и в экспериментах по FRET, в сочетании с жёлтыми флуоресцентными белками, например Венерой (см. рисунок 4), демонстрирует значительный рост отношения сигнал-шум.
Красные флуоресцентные белки
Главным направлением развития флуоресцентных белков стало создание такого красного деривата, которые бы не уступали или даже превосходили усиленные зелёные флуоресцентные белки по их полезным свойствам. Преимуществом красных флуоресцентных белков является их потенциальная совместимость с существующими конфокальными и широкопольными микроскопами (и их фильтрационными наборами), а также высокая эффективность при наблюдении целых животных, которые значительно прозрачнее для красного света. Поскольку создание мутантов из зелёного флуоресцентного белка медузы Aequorea victoria не увенчалось успехом за пределами жёлтого диапазона, внимание исследователей было обращено к тропическим рифовым кораллам.
Первый широко применяемый коралловый флуоресцентный белок был получен из Discosoma striata (Дискосомы полосатой) и обозначается DsRed (DsКрасный). Полностью созревший DsКрасный имеет максимум флуоресценции на 583 нанометрах и два пика в спектре возбуждения: основной на 558 нанометрах и другой, ниже, на 500 нанометрах. Тем не менее, использование DsКрасного сопряжено с некоторыми проблемами. Созревание флуоресценции DsКрасного происходит медленно и проходит через стадию зелёного флуоресцентного свечения. Называемый зелёным состоянием, этот артефакт оказался весьма проблематичным в экспериментах по мультиокрашиванию с применением других зелёных флуоресцентных белков из-за возникающего спектрального перекрытия. Более того, DsКрасный является тетрамером и может образовывать в живых клетках большие белковые соединения. И хотя эти свойства несущественны при использовании DsКрасного в качестве репортерной группы экспрессии генов, его эффективность как метки антигенной детерминанты крайне ограничена. В отличии от флуоресцентных белков медузы, с помощью которых были помечены сотни белков, DsКрасный оказался гораздо менее успешным, а часто и токсичным.
Некоторые проблемы DsКрасных флуоресцентных белков были преодолены с помощью мутагенеза. Второе поколение DsКрасных, известное как DsRed2 (DsКрасный2), несёт в себе несколько мутаций в пептидно-аминном сайте терминации транскрипции, которые предотвращают формирование белковых групп и сокращают токсичность. К тому же, при этих модификациях сокращается время созревания флуророфора. Белок DsКрасный2 по-прежнему образует тетрамер, но более сочетаемый с зелёными флуоресцентными белками в экспериментах по мультиокрашиванию благодаря более быстрому созреванию. Дальнейшее сокращение времени созревания было реализовано в третьем поколении DsКрасных мутантов, которые также демонстрировали повышенную яркость по сравнению с пиком клеточной флуоресценции. Красное флуоресцентное свечение белка DsRed-Express (DsКрасный-Экспресс) наблюдается уже через час после экспрессии, в сравнении с 6 часами для DsКрасного2 и 11 часами для DsКрасного. Был также разработан вариант белка, оптимизированный для дрожжей, под названием RedStar (Красная звезда), который тоже отличается ускоренным созреванием и повышенной яркостью. Поскольку присутствие зелёного состояния в DsКрасном-Экспрессе и Красной звезде не явно, эти флуоресцентные белки являются наилучшими вариантами в оранжево-красном диапазоне в экспериментах по мультиокрашиванию. К сожалению, эти красители всегда образуют тетрамеры, поэтому они не являются самыми лучшими метками для белков.
Рис. 5. Спектральные кривые оранжевых и красных мономерных флуоресцентных белков
Из организмов рифовых кораллов было выделено несколько дополнительных красных флуоресцентных белков, оказавшихся весьма перспективными. Одним из первых, применённых к клеткам млекопитающих, был HcRed1 (HcКрасный1), который был выделен из Heteractis crispa и сейчас выпускается серийно. HcКрасный1 был получен из не флуоресцирующего хромопротеина, поглощающего красный свет. В процессе мутагенеза удалось произвести слабо флуоресцирующий облигатный (обязательный) димер с максимумом поглощения на 588 нанометрах и максимумом испускания на 618 нанометрах. Хотя флуоресцентный спектр этого белка вполне отличим от спектра DsКрасного, он склонен соединятся с последним, являясь гораздо менее ярким. Для преодоления димеризации была разработана интересная конструкция из двух тандемно соединённых HcКрасных2 белков, которая могла бы служить в качестве мономерной метки. Но поскольку общая яркость двойного белка не повысилась, он не стал подходящим вариантом для стандартных приложений в микроскопии живых клеток.
Развитие мономерных вариантов флуоресцентных белков
В естественных условиях большинство флуоресцентных белков существуют в качестве димеров, тетрамеров и олигомеров более высокого порядка. Предполагается, например, что зелёный флуоресцентный белок медузы Aequorea victoria, является частью тетрамерного комплекса с экварином, правда это явление наблюдалось лишь при очень высоких концентрациях белка, и склонность флуоресцентных белков медузы образовывать димеры, в общем, очень слаба (константа диссоциации более 100 микромолей). Таким образом, при экспрессии флуоресцентных белков в тканях млекопитающих их димеризация обычно не наблюдается. Тем не менее, при нацеливании флуоресцентных белков в определённые клеточные компартменты, например, в плазматическую мембрану, местная концентрация белков, теоретически, может быть достаточна для димеризации. Это необходимо учитывать особенно при проведении FRET экспериментов, в которых сложные наборы данных могут быть испорчены артефактами вследствие димеризации.
Создание мономерных вариантов DsКрасного оказалось трудной задачей. Более 30 аминокислотных замен в структуре белка потребовалось для создания мономерного белка DsКрасный первого поколения (называемого RFP1). Однако, этот дериват демонстрировал очень слабую флуоресценцию в сравнении с исходным белком и очень быстро фотообесцвечивался, благодаря чему он был гораздо менее эффективен по сравнению с зелёными и жёлтыми флуоресцентными белками. Чтобы ослабить тенденцию потенциально эффективных биологических зондов сливаться друг с другом и сдвинуть максимумы свечения в сторону длинных волн, не прекращаются исследования по мутагенезу жёлтых, оранжевых, красных и тёмно-красных вариантов флуоресцентных белков, включая новейшие методы, например, соматичесую сверхмутацию.
Были получены мономерные флуоресцентные белки с высоким коэффициентом поглощения, квантовым выходом и фотостабильностью, хотя ни один вариант до сих пор не является оптимальным по всем критериям одновременно. В результате усилий по преодолению обязательной тетрамерности красных флуоресцентных белков, осложняющей экспрессию, были получены мономерные варианты, более приспособленные к биологическому функционированию.
Возможно самым впечатляющим достижением в этом направлении явилось создание нового поколения флуоресцентных белков, полученных из мономерного красного флуоресцентного белка путём направленного мутагенеза остатков Q66 и Y67. Белки этой группы, названные по фруктам, цвета которых совпадают с цветом флуоресценции красителей (см. таблицу 1 и рисунок 5), имеют максимум свечения в диапазоне от 560 до 610 нанометров.
Дальнейшим расширением этого класса многократной соматической сверхмутацией явились флуоресцентные белки с испусканием на длинах волн до 650 нанометров. Эти новые белки в значительной мере заполнили пробел между самыми красными флуоресцентными белками из медузы (такими как Венера) и красными флуоресцентными белками из рифовых кораллов. Хотя многим из этих новых белков не хватает яркости и стабильности, необходимых во многих экспериментах, само их существование обнадёживает и воодушевляет исследователей на создание ярких стабильных мономерных флуоресцентных белков во всём видимом диапазоне.
Оптические маркеры
Одним из наиболее интересных направлений является применение флуоресцентных белков в качестве молекулярных или оптических маркеров (см. таблицу 1), меняющих цвет или интенсивность свечения в результате внешнего светового воздействия или с течением времени. Например, в результате точечной мутации исходного пептида медузы получается фотоактивируемый вариант зелёного флуоресцентного белка (известный как PA-GFP). При облучении его светом из диапазона около 400 нанометров происходит его фотоконверсия, при которой пик возбуждения сдвигается из ультрафиолета в синий диапазон. Спектральная кривая неконвертированного PA-GFP аналогичен профилю исходного белка (с пиком возбуждения приблизительно от 395 до 400 нанометров). После фотоконверсии пик возбуждения на 488 нанометрах возрастает приблизительно в 100 раз. В результате этого возникает очень резкий контраст между неконвертированными и конвертированными пулами белков PA-GFP, что оказывается эффективным для прослеживания динамики субпопуляций молекул внутри клеток. На рисунке 6(а) представлено изображение живой трансфектированной клетки млекопитающего до фотоконверсии белков PA-GFP, находящихся в её цитоплазме и возбуждаемых аргоновым ионным лазером на 488 нанометрах. Изображение той же клетки, но уже после фотоконверсии PA-GFP, осуществлённой синим диодным лазером на длине 405 нанометров, представлено на рисунке 6(d).
Табл. 1. Свойства отдельных оптических маркеров
|
Белок (акроним) |
Максимум возбуждение (нм) |
Максимум испускание (нм) |
Коэффициент молярной экстинкции |
Квантовый выход |
Структура in vivo |
Относительная яркость (%отEGFP) |
|
PA-GFP |
504 |
517 |
17,400 |
0.79 |
Мономер |
41 |
|
CoralHue Kaede (G) |
508 |
518 |
98,800 |
0.88 |
Тетрамер |
259 |
|
CoralHue Kaede (R) |
572 |
580 |
60,400 |
0.33 |
Тетрамер |
59 |
|
CoralHue Dronpa (G) |
503 |
518 |
95,000 |
0.85 |
Мономер |
240 |
|
Kindling (KFP1) |
580 |
600 |
59,000 |
0.07 |
Тетрамер |
12 |
|
PS-CFP (C) |
402 |
468 |
34,000 |
0.16 |
Мономер |
16 |
|
PS-CFP (G) |
490 |
511 |
27,000 |
0.19 |
Мономер |
15 |
|
mEosFP (G) |
505 |
516 |
67,200 |
0.64 |
Мономер |
128 |
|
mEosFP (O) |
569 |
581 |
37,000 |
0.62 |
Мономер |
68 |
Другие флуоресцентные белки также могут использоваться в качестве оптических маркеров. Трёхфотонным возбуждением (на длине волны не более 760 нанометров) флуоресцентного белка DsКрасного его красное свечение можно конвертировать в зелёное. Этот эффект связан, вероятно, с избирательным фотообесцвечиванием красных хромофоров в DsКрасном, благодаря чему становится заметной флуоресценция в зелёном состоянии. Вариант DsКрасного, называемый Таймером (Timer) постепенно, в течение нескольких часов, меняет своё свечение с ярко зелёного (500 нанометров) на ярко красное (580 нанометров). Соотношение зелёной и красной флуоресценции может быть использовано для сбора временны?х данных в исследованиях экспрессии генов.
Фотопереключаемый оптический маркер PS-CFP получен мутагенезом варианта зелёного флуоресцентного белка. При освещении этого белка на 405 нанометрах его флуоресценция меняется с голубой на зелёную (этот вид фотоконверсии представлен на рисунках 6 (b) и (e)). Будучи мономером, этот зонд может быть особенно эффективен при изучении фотообесцвечивания, фотоконверсии и фотоактивации. Однако, яркость свечения PS-CFP примерно в 2,5 раза ниже, чем у PA-GFP, и, по сравнению с другими маркерами, его фотоконвертируемость не так высока (40-нанометровый сдвиг флуоресценции при фотоконверсии меньше, чем наблюдаемый у аналогичных красителей). Дополнительный мутагенез этого или «родственных» ему флуоресцентных белков, возможно, приведёт к более эффективным вариантам в этом спектральном диапазоне.
Рис. 6. Флуоресцентные белки-маркеры
Оптические маркеры были также получены из флуоресцентных белков, клонированных из кораллов и актиний. Флуоресцентный белок Kaede, выделенный из мадрепорового (каменистого) коралла, в ультрафиолетовом свете фотоконвертируется из зелёного в красный. В отличии от PA-GFP, конверсия флуоресценции белка Kaede возникает при поглощении света, спектрально отличного от того, на котором происходит возбуждение. К сожалению, этот белок является облигатным тетрамером, что делает его менее удобным в качестве метки антигенной детерминанты, по сравнению с PA-GFP. Другой тетрамерный вариант флуоресцентного белка мадрепорового коралла (Lobophyllia hemprichii), называемый EosFP (см. таблицу 2), при облучении ультрафиолетом приблизительно на 390 нанометрах меняет ярко зелёную флуоресценцию на оранжево-красную. В этом случае спектральный сдвиг происходит в результате фотоиндуцированной модификации, заключающейся в разрыве полипептидного остова, прилегающего к хромофору. В результате дальнейшего мутагенеза «дикого» белка EosFP были получены мономерные дериваты, которые могут оказаться полезными в создании гибридных белков.
Третьим оптическим маркером, полученным не из Aequorea, является разжигаемый (Kindling) флуоресцентный белок (KFP1), созданный на основе нефлуоресцентного хромопротеина, выделенного из Anemonia sulcata (сейчас он серийно выпускается фирмой «Евроген»). Разжигаемый флуоресцентный белок не светится, пока не облучён зелёным светом. Свет низкой интенсивности вызывает кратковременное красное свечение, которое затухает через несколько минут (см. митохондрии на рисунке 6©). Облучение синим светом немедленно тушит «разожжённую» флуоресценцию, позволяя тем самым надёжно контролировать флуоресцентное окрашивание. И напротив, высоко интенсивное освещение вызывает необратимое разжигание, приводящее к стабильной подсветке (окрашиванию), подобно белку PA-GFP (рисунок 6(f)). Возможность точного управления флуоресценцией особенно важна при слежении за движением частиц в плотном окружении. Этот метод, например, позволяет проследить развитие клеток нервной пластинки эмбриона Xenopus (шпорцевой лягушки) и движение отдельных митохондрий в клетках PC12.
Развитие оптических маркеров продолжается, и оно идёт в направлении создания эффективных для молекулярного маркирования флуоресцентных белков, их более ярких мономерных вариантов, легко фотоковертируемых и представляющих широкий спектр цветов. Эти достижения, а также микроскопы, оснащённые оборудованием, позволяющем гармонично сочетать режимы освещения для наблюдения флуоресценции и направленную маркировку, станут обычными для биологических лабораторий в не столь отдалённом будущем. В конце концов, эти инновации способны привести к важным достижениям в исследовании пространственно-временной динамики систем сигнальной трансдукции.
Векторы флуоресцентных белков и перенос генов
Флуоресцентные белки достаточно универсальны и успешно применяются почти во всех биологических дисциплинах от микробиологии до системной физиологии. Эти красители уже стали повсеместными и успешно применяются в качестве репортерных молекул в изучении экспрессии генов в культивируемых клетках и тканях и живых организмах. В живых клетках флуоресцентные белки обычно используются для наблюдения местоположения и динамики белков, органелл и других клеточных компартментов. Было разработано много различных методик для гибридизации флуоресцентных белков и усиления их экспрессии в клетках млекопитающих и других системах. Основным средством доставки химерных генетических последовательностей флуоресцентных белков в клетки являются бактериальные плазмиды и вирусные векторы, полученные методами генной инженерии.
Гены флуоресцентных белков могут быть введены в клетки млекопитающих и другие клетки с помощью соответствующих временных или стабильных векторов (обычно это плазмиды или вирусы). В экспериментах по временной экспрессии генов (часто называемых врeменной трансфекцией) ДНК плазмид или вирусов, введённых в организм-хозяин, не обязательно сливаются с хромосомами, но могут экспрессироваться в цитоплазме в течении короткого периода времени. Экспрессия слитых (гибридных) генов, легко отслеживаемая по флуоресцентному свечению с помощью фильтрационных блоков, соответствующих данному флуоресцентному белку, начинается в течении нескольких часов после трансфекции и продолжается от 72 до 96 часов после введения ДНК плазмид в клетки млекопитающих. Во многих случаях ДНК плазмид могут быть введены в геном устойчивым образом, что порождает стабильные трансформированные клеточные линии. Выбор между временной и стабильной трансфекцией зависит от объектов-мишеней в данном эксперименте.
Рис. 7. Рестрикционный фермент и генетическая карта серийно выпускаемого деривата бактериальной плазмиды
Базовая конфигурация плазмидного вектора, способного обеспечить перенос генов флуоресцентного белка, имеет несколько необходимых компонентов. Плазмиды должны содержать нуклеотидные последовательности прокариотной клетки, кодирующие начало репликации ДНК в бактерии, и ген устойчивости к антибиотику. Эти элементы, часто называемые челночными последовательностями, обеспечивают размножение и отбор плазмид в бактерии-хозяине для производства необходимого количества векторов для трансфекции в клетки млекопитающих. В дополнение, плазмиды должны содержать один или несколько эукариотических генетических элементов для управления началом транскрипции информационной РНК, сигнал полиаденилирования в клетках млекопитающих, интрон (необязательно) и ген для ко-селекции в клетках млекопитающих. Транскрипционные элементы необходимы для экспрессии представляющего интерес гибридного белка в клетке-хозяине млекопитающего, а селективным геном обычно является антибиотик, обеспечивающий устойчивость содержащих плазмиды клеток. Эти общие условия могут меняться в зависимости от назначения плазмид, и многие векторы имеют широкий спектр дополнительных компонентов, необходимых для конкретных приложений.
На рисунке 7 представлен рестрикционный фермент и генетическая карта серийно выпускаемого (БД Биосайенсис Клонтек) деривата бактериальной плазмиды, содержащей кодирующую последовательность усиленного жёлтого флуоресцентного белка, слитого с последовательностью выступающего в качестве мишени калретикулина (резидентного белка) эндоплазматической сети. В результате экспрессии этого генного продукта в восприимчивых клетках млекопитающих возникает содержащий EYFP химерный пептид, локализованный в мембране эндоплазматической сети и специально разработанный для флуоресцентного окрашивания этой органеллы. Вектор-хозяин является дериватом высококопийной плазмиды pUC (приблизительно 500 копий), содержащей бактериальный репликатор, обеспечивающий размножение специальных штаммов E. сoli. Ген антибиотика канамицина, экспрессированный в бактерии, сообщает ей устойчивость в качестве селективного маркера.
Вектор EYFP, представленный выше, дополнительно содержит промотор цитомегаловируса (CMV) человека для обеспечения экспрессии генов в линии трансфектированных клеток человека и млекопитающих, а также репликатор бактериофага f1 для производства однонитевых ДНК. Векторный остов также включает в себя репликатор обезьяньего вируса (ОВ-40), активного в клетках млекопитающих, которые экспрессируют Т-антиген вируса ОВ-40. Селекция устойчивых к антибиотику G418 трансфектантов обеспечивается кассетой, кодирующей резистентность к неомицину и состоящей из раннего промотора ОВ-40, гена устойчивости к неомицину (аминогликозид 3′-фосфотрансферазы) и сигналов полиаденилирования для вируса простого герпеса тимидинкиназы (ТК ВПГ) для стабильности мРНК. Для большей степени универсальности плазмиды в её остове (см. рисунок 7) присутствуют шесть уникальных рестрикционных ферментов.
Размножение, выделение и трансфекция плазмид флуоресцентных белков
Успешная трансфекция в клетках млекопитающих зависит от того, насколько чистыми и свободными от бактериальных эндотоксинов будут применяемые в эксперименте плазмидные или вирусные векторы. В нативном состоянии молекула кольцевой плазмидной ДНК представляет собой третичную сверхспиральную структуру, в которой двойная спираль закручена вокруг себя несколько раз. В течение лет методом очистки сверхспиральных плазмидных и вирусных ДНК было центрифугирование в градиенте плотности хлористого цезия в присутствии интеркалирующего агента (например, бромистого этидия или йодида пропидия). Этим методом, затратным с точки зрения как оборудования, так и материалов, сверхспиральная (плазмидная) ДНК разделяется на линейную хромосомную и разорванную кольцевую ДНК в соответствии с плавучей плотностью, позволяя собирать плазмидные ДНК высокой степени очистки. Недавно, этот громоздкая и долгая процедура центрифугирования была заменена упрощённым методом ионообменной хроматографии (обычно называемым mini-prep), позволяющим получать свободные от эндотоксинов плазмидные ДНК относительно быстро и в больших количествах.
Специальные бактериальные мутанты, называемые компетентными клетками, были разработаны для удобного и относительно недорогого умножения плазмидных векторов. Благодаря набору специальных мутаций эти бактерии особенно восприимчивы к репликации плазмид, а химически нарушенная проницаемость их мембраны обеспечивает перенос (называемый трансформацией) ДНК через саму мембрану и клеточную оболочку. После трансформации бактерии выращиваются до логарифмической фазы в присутствии антибиотика, с помощью которого осуществляется отбор, управляемый плазмидами с кодом этого антибиотика. Бактериальная культура уплотняется центрифугированием и лизируется раствором щелочного моющего средства, содержащего ферменты для очистки от РНК. После этого лизат фильтруется и помещается на ионообменную колонку. Нежелательные компоненты, включая РНК, ДНК и белки, тщательно вымываются из колонки перед тем, как ДНК плазмид извлекаются с помощью высокосолевого буфера. После этого извлечённые ДНК плазмид осаждаются в спиртовом растворе (изопропаноле), собираются центрифугированием, промываются и опять растворяются в буфере. Очищенные таким образом ДНК готовы для экспериментов по трансфекции.
Рис. 8. Липидная трансфекция в клетки млекопитающих
Клетки млекопитающих, используемые для трансфекции, должны быть в превосходном физиологическом состоянии и находиться в логарифмической фазе роста. Для оптимизации внедрения плазмидных ДНК в культивируемые клетки были разработаны многочисленные трансфекционные реагенты, выпускаемые сегодня серийно. Методики внедрения разнообразны: от простого осаждения фосфатом кальция до связывания плазмидных ДНК липосомами, которые проникая сквозь клеточную мембрану, доставляют содержимое в цитоплазму (как показано на рисунке 8). Обобщённо называемые липофекцией, методы с использованием липидов получили широкое распространение благодаря своей эффективности для большого количества популярных клеточных линий и применяются сегодня в большинстве экспериментов по трансфекции.
При врeменной трансфекции потеря плазмидного гена происходит обычно в течение относительно короткого периода (несколько дней), в то время как стабильные линии трансфектированных клеток вырабатывают «гостевые» белки достаточно долго (от нескольких месяцев до нескольких лет). Стабильные клеточные линии могут отбираться с помощью антибиотических маркеров в плазмидном остове (см. рисунок7). Одним из наиболее распространённых антибиотиков для селекции стабильных трансфектантов в линиях клеток млекопитающих является лекарственный препарат G418, подавляющий синтез белков, не его доза значительно меняется в зависимости от клеточной линии. Для селекции стабильных клеток были разработаны и другие часто применяемые антибиотики, включая гидромицин B и пуромицин, которые тоже имеют генетические маркеры. Эффективность получения стабильных клеточных линий определяется эффективностью начальной трансфекции. Электропорация, в этом отношении, оказалась надёжным способом получения стабильных трансфектантов линеаризованными плазмидами и очищенными генами. Метод электропорации заключается в пропускании коротких импульсов высокого напряжения через клеточную суспензию, в результате чего в плазматических мембранах образуются поры, через которые трансфекционные ДНК попадают в клетки. Процедура электропорации требует специального оборудования, тем не менее, его стоимость сравнима с затратами на липофекционные реагенты при выполнении большого числа трансфекций.
Будущее флуоресцентных белков
Современное развитие флуоресцентных белков идёт по двум основным направлениям. Первое — окончательная «доводка» существующей палитры (от синих до жёлтых) флуоресцентных белков, полученных из медузы Aequorea victoria, а второе — усовершенствование мономерных флуоресцентных белков, испускающих в видимом диапазоне от оранжевого до дальнего красного. Прогресс, достигнутый в этих направлениях, действительно впечатляет, и появление на горизонте ближних инфракрасных флуоресцентных белков уже не кажется невообразимым.
Последнее поколение белков, выделенных из медузы, во многом восполнило дефицит первых флуоресцентных белков, особенно жёлтых и зелёных дериватов. Поиск мономерного, яркого и быстро созревающего красного флуоресцентного белка привёл к нескольким новым и интересным классам флуоресцентных белков, особенно тех, которые были выделены из кораллов. Дальнейшее расширение цветовой палитры происходит как за счёт развития существующих флуоресцентных белков, так и благодаря новым технологиям, например, внедрению искусственных аминокислот. А поскольку оптические методы спектрального разделения продолжают развиваться и становятся более распространёнными, эти новые варианты обязательно дополнят существующую палитру, особенно в жёлтом и красном участках спектра.
Сегодня в технологии флуоресцентных зондов имеет место тенденция к повышению роли красителей, флуоресцирующих в дальнем красном и ближнем инфракрасном. В клетках млекопитающих и автофлуоресценция, и поглощение света резко сокращаются на красном конце спектра. Таким образом, развитие дальних красных флуоресцентных красителей будет чрезвычайно полезным для исследования толстых образцов и животных целиком. В случае успешного использования флуоресцентных белков в качестве репортерных молекул в трансгенных системах, применение красных флуоресцентных белков в целых организмах станет в ближайшие годы чрезвычайно важным.
Наконец, только сейчас начинает осознваться огромный потенциал приложения флуоресцентных белков для создания биосенсоров. Количество различных биосенсоров стремительно растёт. Использование структурной информации приводит к постоянному повышению их чувствительности. Успехи в этом направлении вселяют уверенность в том, что почти любой биологический параметр можно будет измерить биосенсором на базе соответствующего флуоресцентного белка.
§ 9. Физико-химические свойства белков
§ 9. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА БЕЛКОВ
Белки – это очень крупные молекулы, по своим размерам они могут уступать только отдельным представителям нуклеиновых кислот и полисахаридам. В таблице 4 представлены молекулярные характеристики некоторые белков.
Таблица 4
Молекулярные характеристики некоторых белков
|
Белок
|
Относитель-ная молекулярная масса
|
Число цепей
|
Число аминокислотных остатков
|
|
Инсулин
|
5733
|
2
|
51
|
|
Рибонуклеаза
|
13683
|
1
|
124
|
|
Миоглобин
|
16890
|
1
|
153
|
|
Химотрипсин
|
22600
|
3
|
241
|
|
Гемоглобин
|
64500
|
4
|
574
|
|
Глутамат-дегидрогеназа
|
~1000000
|
~40
|
~8300
|
В молекулах белков может содержаться самое разное количество аминокислотных остатков — от 50 и до нескольких тысяч; относительные молекулярные массы белков также сильно колеблются — от нескольких тысяч (инсулин, рибонуклеаза) до миллиона (глутаматдегидрогеназа) и более. Число полипептидных цепей в составе белков может составлять от единицы до нескольких десятков и даже тысяч. Так, в состав белка вируса табачной мозаики входит 2120 протомеров.
Зная относительную молекулярную массу белка, можно приблизительно оценить, какое число аминокислотных остатков входит в его состав. Средняя относительная молекулярная масса аминокислот, образующих полипептидную цепь, равна 128. При образовании пептидной связи происходит отщепление молекулы воды, следовательно, средняя относительная масса аминокислотного остатка составит 128 – 18 = 110. Используя эти данные, можно подсчитать, что белок с относительной молекулярной массой 100000 будет состоять приблизительно из 909 аминокислотных остатков.
Электрические свойства белковых молекул
Электрические свойства белков определяются присутствием на их поверхности положительно и отрицательно заряженных аминокислотных остатков. Наличие заряженных группировок белка определяет суммарный заряд белковой молекулы. Если в белках преобладают отрицательно заряженные аминокислоты, то его молекула в нейтральном растворе будет иметь отрицательный заряд, если преобладают положительно заряженные – молекула будет иметь положительный заряд. Суммарный заряд белковой молекулы зависит и от кислотности (рН) среды. При увеличении концентрации ионов водорода (увеличении кислотности) происходит подавление диссоциации карбоксильных групп:
и в то же время увеличивается число протонированных амино-групп;
.
Таким образом, при увеличении кислотности среды происходит уменьшение на поверхности молекулы белка числа отрицательно заряженных и увеличение числа положительно заряженных групп. Совсем другая картина наблюдается при снижении концентрации ионов водорода и увеличении концентрации гидроксид-ионов. Число диссоциированных карбоксильных групп возрастает
и снижается число протонированных аминогрупп
.
Итак, изменяя кислотность среды, можно изменить и заряд молекулы белка. При увеличении кислотности среды в молекуле белка снижается число отрицательно заряженных группировок и увеличивается число положительно заряженных, молекула постепенно теряет отрицательный и приобретает положительный заряд. При снижении кислотности раствора наблюдается противоположная картина. Очевидно, что при определенных значениях рН молекула будет электронейтральной, т.е. число положительно заряженных групп будет равно числу отрицательно заряженных групп, и суммарный заряд молекулы будет равен нулю (рис. 14).
Значение рН, при котором суммарный заряд белка равен нулю, называется изоэлектрической точкой и обозначается pI.
Рис. 14. В состоянии изоэлектрической точки суммарный заряд молекулы белка равен нулю
Изоэлектрическая точка для большинства белков находится в области рН от 4,5 до 6,5. Однако есть и исключения. Ниже приведены изоэлектрические точки некоторых белков:
|
Белок
|
pI
|
|
Пепсин
|
1,0
|
|
Каталаза
|
5,1
|
|
Рибонуклеаза
|
7,8
|
|
Лизоцим
|
11,0
|
При значениях рН ниже изоэлектрической точки белок несет суммарный положительный заряд, выше – суммарный отрицательный.
В изоэлектрической точке растворимость белка минимальна, так как его молекулы в таком состоянии электронейтральны и между ними нет сил взаимного отталкивания, поэтому они могут «слипаться» за счет водородных и ионных связей, гидрофобных взаимодействий, ван-дер-ваальсовых сил. При значениях рН, отличающихся от рI, молекулы белка будут нести одинаковый заряд — либо положительный, либо отрицательный. В результате этого между молекулами будут существовать силы электростатического отталкивания, препятствующие их «слипанию», растворимость будет выше.
Растворимость белков
Белки бывают растворимые и нерастворимые в воде. Растворимость белков зависит от их структуры, величины рН, солевого состава раствора, температуры и других факторов и определяется природой тех групп, которые находятся на поверхности белковой молекулы. К нерастворимым белкам относятся кератин (волосы, ногти, перья), коллаген (сухожилия), фиброин (щелк, паутина). Многие другие белки растворимы в воде. Растворимость определяется наличием на их поверхности заряженных и полярных группировок (-СОО—, -NH3+, -OH и др.). Заряженные и полярные группировки белков притягивают к себе молекулы воды, и вокруг них формируется гидратная оболочка (рис. 15), существование которой обусловливает их растворимость в воде.
Рис. 15. Образование гидратной оболочки вокруг молекулы белка.
На растворимость белка влияет наличие нейтральных солей (Na2SO4, (NH4)2SO4 и др.) в растворе. При малых концентрациях солей растворимость белка увеличивается (рис. 16), так как в таких условиях увеличивается степень диссоциации полярных групп и экранируются заряженные группы белковых молекул, тем самым снижается белок-белковое взаимодействие, способствующее образованию агрегатов и выпадению белка в осадок. При высоких концентрациях солей растворимость белка снижается (рис. 16) вследствие разрушения гидратной оболочки, приводящего к агрегации молекул белка.
Рис. 16. Зависимость растворимости белка от концентрации соли
Существуют белки, которые растворяются только в растворах солей и не растворяются в чистой воде, такие белки называют глобулины. Существуют и другие белки – альбумины, они в отличие от глобулинов хорошо растворимы в чистой воде.
Растворимость белков зависит и от рН растворов. Как мы уже отмечали, минимальной растворимостью обладают белки в изоэлектрической точке, что объясняется отсутствием электростатического отталкивания между молекулами белка.
При определенных условиях белки могут образовывать гели. При образовании геля молекулы белка формируют густую сеть, внутреннее пространство которой заполнено растворителем. Гели образуют, например, желатина (этот белок используют для приготовления желе) и белки молока при приготовлении простокваши.
На растворимость белка оказывает влияние и температура. При действии высокой температуры многие белки выпадают в осадок вследствие нарушения их структуры, но об этом более подробно поговорим в следующем разделе.
Денатурация белка
Рассмотрим хорошо нам знакомое явление. При нагревании яичного белка происходит постепенное его помутнение, и затем образуется твердый сгусток. Свернувшийся яичный белок – яичный альбумин – после охлаждения оказывается нерастворимым, в то время как до нагревания яичный белок хорошо растворялся в воде. Такие же явления происходят и при нагревании практически всех глобулярных белков. Те изменения, которые произошли при нагревании, называются денатурацией. Белки в естественном состоянии носят название нативных белков, а после денатурации — денатурированных.
При денатурации происходит нарушение нативной кон-формации белков в результате разрыва слабых связей (ион-ных, водородных, гидрофобных взаимодействий). В результате этого процесса могут разрушаться четвертичная, третичная и вторичные структуры белка. Первичная структура при этом сохраняется (рис. 17).
Рис. 17. Денатурация белка
При денатурации гидрофобные радикалы аминокислот, находящиеся в нативных белках в глубине молекулы, оказываются на поверхности, в результате создаются условия для агрегации. Агрегаты белковых молекул выпадают в осадок. Денатурация сопровождается потерей биологической функции белка.
Денатурация белка может быть вызвана не только повышенной температурой, но и другими факторами. Кислоты и щелочи способны вызвать денатурацию белка: в результате их действия происходит перезарядка ионогенных групп, что приводит к разрыву ионных и водородных связей. Мочевина разрушает водородные связи, следствием этого является потеря белками своей нативной структуры. Денатурирующими агентами являются органические растворители и ионы тяжелых металлов: органические растворители разрушают гидрофобные связи, а ионы тяжелых металлов образуют нерастворимые комплексы с белками.
Наряду с денатурацией существует и обратный процесс – ренатурация. При снятии денатурирующего фактора возможно восстановление исходной нативной структуры. Например, при медленном охлаждении до комнатной температуры раствора восстанавливается нативная структура и биологическая функция трипсина.
Белки могут денатурировать и в клетке при протекании нормальных процессов жизнедеятельности. Совершенно очевидно, что утрата нативной структуры и функции белков – крайне нежелательное событие. В связи с этим следует упомянуть об особых белках – шаперонах. Эти белки способны узнавать частично денатурированные белки и, связываясь с ними, восстанавливать их нативную конформацию. Шапероны также узнают белки, процесс денатурации которых зашел далеко, и транспортируют их в лизосомы, где происходит их расщепление (деградация). Шапероны играют важную роль и в процессе формирования третичной и четвертичной структур во время синтеза белка.
Интересно знать! В настоящее время часто упоминается такое заболевание, как коровье бешенство. Эту болезнь вызывают прионы. Они могут вызывать у животных и человека и другие заболевания, носящие нейродегенеративный характер. Прионы – это инфекционные агенты белковой природы. Прион, попадая в клетку, вызывает изменение конформации своего клеточного аналога, который сам становится прионом. Так возникает заболевание. Прионный белок отличается от клеточного по вторичной структуре. Прионная форма белка имеет в основном b-складчатую структуру, а клеточная – a-спиральную.
Антигенные свойства рекомбинантного аналога белка легумаин трематоды Opisthorchis felineus, вызывающей описторхоз у человека | Разумов
1. Государственный доклад // Федеральный центр ги-гиены и эпидемиологии Роспотребнадзора. М., 2009. C. 349—350.
2. Котелкин А.Т., Разумов И.А., Локтев В.Б. Получение и характеризация мышиных гибридом, секретирующих моноклональных антитела к основному растворимому антигену Opisthorchis felineus // Мед. паразитология и паразитарные болезни. 1999. T. 3. С. 6—10.
3. Остерман А.А. Методы исследования белков и нуклеиновых кислот. Электрофорез и ультрацентрифугирование. 1981. C. 37—64.
4. Eursitthichai V., Viyanant V., Vichasri-Grams S. et al. Mo-lecular Cloning and Characterisation of a Glutathione S-Transferase Encoding Gene From O.viverrini // Asian Pacific Journal of Allergy and Immunology. 2004. V 22. P. 219—228.
5. Hong S.-J., Yun Kim T., Gan X.-X. et al. Clonorchis sinen-sis: glutathione S-transferase as a serodiagnostic antigen for detecting IgG and IgE antibodies. // Experimental parasitology. 08.2002. V. 101. P. 231—234.
6. Hu F., Yu X. et al. Clonorchis sinensis: expression, charac-terization, immunolocalization and serological reactivity of one excretory/secretory antigen-LPAP homologue // Exp. Parasitol. 2007. V. 117 (2). P. 157—221.
7. Ju J.W., Joo H.N. et al. Identification of a serodiagnostic antigen, legumain, by immunoproteomic analysis of excre-tory-secretory products of Clonorchis sinensis adult worms // Proteomics. V. 9 (11). P. 3066—3144.
8. King S., Scholz T. Trematodes of the family Opisthorchii-dae: a minireview // The Korean Journal of Parasitology. 2001. V. 39. P. 209—221.
9. Laha T., Sripa J., Sripa B. et al. Asparaginal endopeptidase from carcinogenetic liver fluke O.viverrini and its potential for serodiagnosis // Int. J. Infect. Dis. 2008. V. 12 (6). P. e49—e59.
10. Li S., Shin J.G., Cho P.Y. et al. Multiple recombinant anti-gens of C.sinensis for serodiagnosis of human clonorchiasis // Parasitol Res. 2011. V. 108. P. 1295—1302.
11. Sambrook J., Fritsch E. F., Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. 1989. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
12. Shen C., Lee J.A. et al. Serodiagnostic applicability of re-combinant antigens of Clonorchis sinensis expressed by wheat germ cell-free protein synthesis system // Diagn. Microbiol. Infect. Dis. 2009. V. 64 (3). P. 334—343.
13. Sripa B., Kaewkes S., Sithithaworn P. et al. Liver Fluke In-duces Cholangiocarcinoma // PLoS Medicine. 2007. V. 4. P. 1148—1155.
14. Towbin H., Staekelin T., Gordon J. Electrohporetic transfer of proteins from polyacrylamide gels tonitrocellu-lose sheets. Procedure and some applications. // Proc. Natl. Acad. Sci. USA. 1979. V. 76. P. 4350—4354.
15. World Health Organization Study Group. Geneva, 2005. P. 243.
16. Zhao Q.P., Moon S.U. et al. Evaluation of Clonorchis sinensis recombinant 7-kilodalton antigen for serodiagnosis of clonorchiasis // Clin. Diagn. Lab. Immunol. 2004. V. 11 (4). P. 814—821.
Protein Property — обзор
Для этого исследования последовательности белков были получены из UniProt, который является хранилищем свойств белка. BEST1 записан с самой длинной последовательностью 585 а.о., тогда как PRPh3 с самой короткой длиной 346 а.о. Мы рассчитали состав каждой аминокислоты в BEST1 и PRPh3 с помощью статистического анализа белковых последовательностей (SAPS) (Brendel, Bucher, Nourbakhsh, Blaisdell, & Karlin, 1992). Наблюдался больший состав лейцина, за которым следовали пролин, серин и глутамин в BEST1 (рис.6.7А). Точно так же в последовательности PRPh3 мы распознали лагерный состав лейцина, за которым следовали серин, лизин и глицин (фиг. 6.7A). Кроме того, мы также рассчитали склонность каждой аминокислоты в нативном и мутантном состоянии BEST1 и PRPh3 (рис. 6.7B). В BEST1 наиболее распространенной аминокислотой в нативном состоянии был лейцин, в то время как валин был наиболее распространенной заменой в PRPh3 (таблица 6.1). Leu → Val, Val → Met, Asp → Asn, Glu → Lys и Leu → Pro были наиболее частыми заменами из-за SAP.Было предсказано, что восемь замен от Leu → Val и семь замен от Val → Met являются патогенными с помощью PolyPhen-2 и SNPs & GO, а также SIFT, PolyPhen-2 и PANTHER. Шесть замен из Asp → Arg, Glu → Lys и Leu → Pro были предсказаны как патогенные с помощью PolyPhen-2 и I-MUTANT Suite, PhD-SNP и I-MUTANT Suite, а также PolyPhen-2, PhD-SNP, SNAP, SNPs & GO и I-MUTANT Suite соответственно. Для PRPh3 наиболее распространенной аминокислотой дикого типа был Pro, в то время как Arg был наиболее распространенной аминокислотой замещения.R → W, G → D, P → L, R → W, C → Y, G → S и P → R были наиболее частыми заменами из-за SAP. Из таблицы 6.1 мы можем сделать вывод, что появление пяти замен из Arg → Trp и Gly → Asp было предсказано как патогенное с помощью PhD-SNP, PANTHER, SNAP, I-MUTANT Suite и Align-GVGD и PANTHER, SNAP, I- MUTANT Suite и Align-GVGD соответственно. Четыре замены из Pro → Leu были предсказаны PANTHER, I-MUTANT Suite и Align-GVGD как патогенные, а три замены из Cys → Tyr, Gly → Ser и Pro → Arg были предсказаны SIFT, PolyPhen- 2, PhD-SNP, SNAP, I-MUTANT Suite и Align-GVGD; PolyPhen-2, PANTHER, I-MUTANT Suite и Align-GVGD; и PolyPhen-2, PANTHER, SNAP, I-MUTANT Suite и Align-GVGD соответственно.
Рисунок 6.7. (А) Состав аминокислот в BEST1 и PRPh3 путем статистического анализа белковых последовательностей. (B) Состав аминокислот в варианте и положении замещения BEST1 и PRPh3.
Фундаментальное свойство белка, термодинамическая стабильность, выявленное исключительно в результате крупномасштабных измерений функции белка
Abstract
Способность белка выполнять заданную функцию является результатом фундаментальных физико-химических свойств, которые включают структуру белка и механизм действия , и термодинамическая устойчивость.Традиционные подходы к изучению этих свойств обычно требовали прямого измерения интересующего свойства, что зачастую было трудоемким мероприятием. Хотя свойства белка можно исследовать с помощью мутагенеза, этот подход ограничен его низкой пропускной способностью. Последние технологические разработки позволили быстро оценить функцию белка, такую как связывание с лигандом, для многочисленных вариантов этого белка. Здесь мы измеряем способность 47 000 вариантов домена WW связываться с пептидным лигандом и используем эти функциональные измерения для выявления стабилизирующих мутаций без непосредственного анализа стабильности.Наш подход основан на хорошо известной концепции, согласно которой функция белка тесно связана со стабильностью. Функция белков обычно снижается из-за дестабилизирующих мутаций, но это снижение может быть устранено за счет стабилизации мутаций. Основываясь на этом наблюдении, мы представляем потенцирование партнера, показатель, который использует эту способность к спасению для идентификации стабилизирующих мутаций, и идентифицируем 15 кандидатных стабилизирующих мутаций в WW-домене. Мы проверили шесть кандидатов термической денатурацией и обнаружили две высокостабилизирующие мутации, одна более стабилизирующая, чем любая ранее известная мутация.Таким образом, физико-химические свойства, такие как стабильность, скрыты в этих крупномасштабных функциональных данных белка и могут быть выявлены с помощью систематического анализа. Такой подход должен позволить обнаружить другие свойства белка.
Последовательность белка определяет его физико-химические свойства, которые включают структуру, термодинамическую стабильность, способность взаимодействовать с другими молекулами и каталитическую способность (1). Эти свойства, в свою очередь, определяют функцию белка.Поскольку последовательность определяет функцию, мутагенез стал фундаментальным инструментом для понимания того, как работают белки. Мутация может повлиять на функцию белка, когда она изменяет одно или несколько свойств белка, таких как его структура, каталитическая активность или стабильность. Понимание механизма, с помощью которого мутация влияет на функцию белка, традиционно требовало специальных анализов для измерения этих свойств (например, термическая денатурация использовалась для измерения стабильности).
Сочетание отбора и высокопроизводительного секвенирования ДНК позволило методам измерения функции большого числа (до миллионов) мутированных версий белка (называемых здесь вариантами) (2⇓ – 4).Эти методы, известные как «глубокое мутационное сканирование» (4), связывают функцию каждого варианта с его распространенностью в популяции вариантов, отобранных для этой функции. Частоты вариантов в популяции измеряются в массе путем высокопроизводительного секвенирования ДНК гена, кодирующего белок. Изменение частоты каждого варианта количественно оценивается путем сравнения частоты каждого варианта до выбора с его частотой после выбора. Обогащение или истощение каждого варианта посредством выбора служит представителем функции варианта; варианты, содержащие высокофункциональные (полезные) мутации, обогащаются после отбора, тогда как варианты, содержащие плохо функциональные (вредные) мутации, истощаются.Глубокое мутационное сканирование позволяет измерять функциональные последствия большого количества вариантов белка параллельно и, следовательно, дает крупномасштабный набор функциональных данных белка. Основываясь исключительно на этом наборе данных, мы представляем анализ для выявления мутаций, которые стабилизируют белок.
Мутации, стабилизирующие белки, важны как для понимания активности белков, так и для успешной инженерии белков. Стабилизирующие мутации в белковых лекарствах, таких как инсулин (5) и антитела (6), и коммерческих ферментах, таких как субтилизин (7), могут предотвратить протеолиз или неправильную укладку, тем самым увеличивая эффективную активность.Белки незначительно стабильны и становятся нефункциональными, если дестабилизируются выше порогового значения. Таким образом, стабильность белка связана с такими показателями функции белка, как каталитическая активность или связывание лиганда (8⇓ – 10). Например, единичная мутация, снижающая стабильность сверх порогового значения, может резко снизить функцию белка. Однако от такой дестабилизирующей мутации можно избавиться путем введения второй стабилизирующей мутации. Полученный белок превышает порог стабильности и, следовательно, имеет повышенную функцию.
Большинство мутаций являются дестабилизирующими, и поэтому в наборе случайно сгенерированных двойных мутантов редкие стабилизирующие мутации обычно сочетаются с дестабилизирующими мутациями. Таким образом, мы предположили, что можем идентифицировать стабилизирующие мутации на основе их способности спасать многие другие (в основном дестабилизирующие) мутации. Эта гипотеза поднимает интригующую возможность того, что фундаментальные физико-химические свойства определенных вариантов белка (например, вариантов с наивысшей стабильностью) могут быть выведены исключительно из крупномасштабных измерений функции белка.Здесь мы показываем, что систематический анализ этих измерений для большого количества вариантов белка может быть использован для расчета потенцирования партнера, показателя, который выявляет стабилизирующие мутации.
Результаты
Глубокое мутационное сканирование домена WW.
Мы использовали глубокое мутационное сканирование для измерения способности 47 000 уникальных вариантов WW-домена hYAP65 связываться с их полипролиновым пептидным лигандом (2, 4). WW домены опосредуют межбелковые взаимодействия, имеют четко определенную структуру и складываются посредством механизма двух состояний, упрощая последующие измерения термодинамической стабильности (11-13).Мы отобразили библиотеку вариантов WW-домена hYAP65 на поверхности бактериофага Т7. Библиотека была создана путем синтеза допированных олигонуклеотидов, при этом каждый член библиотеки содержал в среднем две мутации в вариабельной области из 102 оснований, кодирующей 34 аминокислоты, которые охватывают большую часть домена. Библиотеку подвергали трем раундам отбора на связывание с биотинилированной формой пептидного лиганда GTPPPPYTVG, которая была иммобилизована на магнитных стрептавидиновых гранулах. Мы выполнили высокопроизводительное секвенирование ДНК входных данных и библиотек из раундов 1–3, получив не менее 10 миллионов считываний для каждой библиотеки (рис.S1 и Таблица S1).
Мы использовали эти данные высокопроизводительного секвенирования ДНК, чтобы получить функциональную оценку для каждого варианта домена WW в библиотеке на основе частоты варианта в каждом раунде отбора. Частоты вариантов были скорректированы на неспецифический перенос, который происходит, когда нефункциональные варианты переносятся от одного раунда к другому из-за фонового связывания гранул и не полностью эффективной промывки (14, 15). Скорость неспецифического переноса оценивалась по характеристикам вариантов, содержащих стоп-кодоны, поскольку эти варианты должны быть нефункциональными.Из неспецифических скорректированных переносимых частот мы построили линейные модели циклического обогащения для каждого из 47 000 вариантов, присутствующих во входной библиотеке, и для всех трех циклов отбора. Для каждого варианта наклон полученной линии указывает на обогащение или истощение этого варианта во время анализа. Чтобы вычислить функциональную оценку, мы разделили наклон каждого варианта на наклон дикого типа (WT) (рис. S2). Мы использовали границу согласия (наклон R 2 ≥ 0,75), чтобы исключить варианты, которые вели себя хаотично.
Взаимодействие между одиночными и двойными мутантами в домене WW.
Мы предположили, что стабилизирующие мутации могут быть обнаружены на основе их способности спасать другие мутации, большинство из которых являются дестабилизирующими. В нашем наборе данных о функциях белков эффект спасения будет наблюдаться, когда две одиночные мутации объединятся в дважды мутированный вариант, чтобы произвести неожиданный функциональный прирост. Эти неожиданные функциональные улучшения, возникающие в результате комбинаций единичных мутаций, можно описать с точки зрения эпистаза (9, 10, 16, 17).Здесь мы определяем эпистаз как возникновение, когда две одиночные мутации ( a и b ) объединяются, чтобы воздействовать на функцию белка иначе, чем ожидалось, исходя из их индивидуальных функциональных эффектов и модели взаимодействия. Мы использовали наиболее распространенную модель, называемую моделью взаимодействия продукта, с оценкой эпистаза, рассчитанной как (ур. 1 )
, где W ab представляет функциональную оценку двойного мутанта, а W a и W, b представляют функциональные баллы одиночных мутантов.
Используя модель продукта, мы вычислили 5 010 индивидуальных баллов эпистаза из функциональных баллов 47 000 вариантов. Модель продукта диктует, что произведение двух функциональных баллов с одним мутантом ( W, a • W b ) должно равняться функциональному баллу с двойным мутантом ( W, ab ), если эпистаз отсутствует. . Мы обнаружили, что функциональные баллы с одним мутантом предсказывают функциональные баллы с двойным мутантом с 0 по шкале Пирсона R 2 .67 (Рис.1 A ). В предыдущем исследовании, посвященном изучению вариантов WW-домена hYAP65, которые выжили после шести раундов отбора на связывание пептида (2), мы получили значение 0,68. Таким образом, несмотря на изменение нашего анализа для объединения последовательных раундов отбора, дополнительного секвенирования и обширной фильтрации данных, мы не улучшили прогнозы функциональных баллов двойных мутантов (рис. S2 и таблица S1). Кроме того, мы протестировали логарифмическую, минимальную и аддитивную модели взаимодействия с оценкой эпистаза, рассчитанной как
(рис.1.
Связь между функцией и эпистазом в массивной коллекции двойных мутантов. ( A ) Функциональная оценка 5010 вариантов с двойной мутацией была предсказана на основе функциональных оценок компонентов с однократной мутацией вариантов с использованием модели продукта. Прогнозируемый функциональный балл нанесен на график против наблюдаемого функционального балла, и эти два показателя сильно коррелируют (Пирсона R 2 = 0,67). Для каждого варианта с двойной мутацией линейные модели, использованные для получения функциональной оценки, имели R 2 ≥ 0.75. ( B ) Показатели эпистаза, рассчитанные с использованием модели продукта для 5010 вариантов, наносятся на график против функциональной оценки дважды мутированного варианта. Распределение баллов эпистаза показано на вставке . Пунктирные линии расположены на расстоянии ± 1 стандартное отклонение от среднего.
Ни одна из этих часто используемых моделей эпистаза (18) не привела к улучшенному предсказанию функциональных баллов двойных мутантов (Рис. S3 A – D ). Мы пришли к выводу, что ограничивающим фактором в прогнозировании функциональных баллов двойных мутантов является точность модели, а не качество измерений функции.Эти результаты доказывают, что эпистаз является внутренним свойством домена hYAP65 WW, а не артефактом качества данных или выбора модели.
В рамках модели продукта, которая наиболее точно предсказывала функциональные баллы двойных мутантов, средний балл эпистаза для всех вариантов был близок к нулю, со стандартным отклонением 0,65 и 86% баллов в пределах 1 стандартного отклонения от среднего. Величина эпистаза оценивается по шкале с величиной вариантной функциональной оценки (рис. 1 B ). Этот эффект масштабирования возникает из-за того, что для расчета оценок эпистаза используются необработанные функциональные баллы по вариантам.Следовательно, если два одиночных мутанта и соответствующий двойной мутант имеют низкие функциональные баллы, они не могут дать высокий балл эпистаза. Мы наблюдали как высокие по величине положительные, так и отрицательные показатели эпистаза, но не наблюдали средней популяционной тенденции к положительному или отрицательному эпистазу (рис. 1 B ). Наиболее функциональные двойные мутанты имеют положительную оценку эпистаза, что позволяет предположить, что высокофункциональные мутанты трудно предсказать.
Чтобы получить представление о паттернах эпистаза в домене WW, мы построили сетевое представление, которое показывает, что в некоторых регионах домена WW мутации, приводящие к положительным эпистатическим взаимодействиям, происходят в положениях, которые также содержат мутации, приводящие к отрицательным взаимодействиям. (Рис.2 А ). Оценки эпистаза распределяются неоднородно, что приводит к возникновению горячих точек (отдельные позиции, в которых некоторые мутации имеют много положительных эпистатических взаимодействий, а другие мутации имеют много отрицательных эпистатических взаимодействий) (рис. 2 A и B ). Эти горячие точки возникают в областях высокого эпистаза, которые включают обе петли, а также часть N-конца (Fig. S3 E ) (критерий суммы рангов Вилкоксона, P = 7,85 × 10 −22 ).
Фиг.2.
Один только эпистаз не позволяет надежно идентифицировать стабилизирующие мутации. ( A ) Показано сетевое представление эпистатических взаимодействий между мутациями. Отдельные мутации представлены в виде узлов на графике и окрашены функциональными баллами (красный цвет соответствует мутациям с более высокими функциональными баллами, чем WT, а синий соответствует мутациям с более низкими функциональными баллами, чем WT). Мутации расположены сначала по положению, а затем по алфавиту по окружности графика по часовой стрелке от координаты 12:00 (увеличьте масштаб, чтобы увидеть отдельные мутации).Последовательность WT показана снаружи графика. Положительные и отрицательные эпистатические взаимодействия между мутациями показаны градиентными красными и синими краями, ширина и оттенок которых пропорциональны величине взаимодействия. Положение β-тяжей в WW-домене указано синими стрелками. Для ясности показаны только эпистатические взаимодействия, по крайней мере, на 1 стандартное отклонение от среднего значения. ( B ) Для каждой позиции в домене доля отрицательных оценок эпистаза откладывается на оси x , а доля положительных показателей эпистаза откладывается на оси y .Доли положительных и отрицательных оценок эпистаза коррелируют между позициями ( R = 0,60, P = 8,8 × 10 −5 ). ( C ) Средняя оценка эпистаза каждого из 192 одиночных мутантов, обнаруженных у 10 или более двойных мутантов, наносится на график против функциональной оценки одиночных мутантов для каждой мутации. Известные стабилизирующие (A20R, L30K и D34T) и повышающие активность (K21R и Q35R) мутации выделены красным и синим цветом соответственно.
Идентификация термодинамически стабилизирующих мутаций.
Поскольку стабилизирующие мутации потенциально могут спасти многие дестабилизирующие мутации, простейшая стратегия их поиска будет полагаться на ожидание, что стабилизирующие мутации будут среди наиболее широко представленных мутаций после отбора. В качестве золотого стандарта мы использовали три известные стабилизирующие мутации домена hYAP65 WW (19, 20), присутствующие в наборе данных (A20R, L30K и D34T), которые в соответствии с этим ожиданием должны стать высоко обогащенными. Однако для этих трех мутаций представление после отбора не было полезным предиктором стабильности (рис.S3 F ). Эта стратегия, вероятно, не увенчалась успехом, потому что, хотя мы измерили большое количество (5010) оценок эпистаза, эти оценки представляют лишь небольшую выборку из 211200 возможных оценок эпистаза в 34 позициях, которые варьировались.
Независимо от общего представления после отбора, стабилизирующие мутации должны спасти многие другие, в основном дестабилизирующие мутации. Каждое из этих действий по спасению будет иметь положительную оценку эпистаза. Таким образом, мы предположили, что стабилизирующие мутации должны быть теми редкими мутациями, которые имеют большой положительный средний балл эпистаза.Мы вычислили средний балл эпистаза для каждой одиночной мутации a , который состоит из среднего значения всех показателей эпистаза, полученных от двойных мутантов, содержащих a и любую другую одиночную мутацию b . Однако средний балл эпистаза также не смог отделить все три известные стабилизирующие мутации домена hYAP65 WW, присутствующие в нашем наборе данных, от основной массы мутаций, а также двух известных мутаций, повышающих активность (19–22) (рис. 2 С ). Средняя оценка эпистаза не смогла правильно идентифицировать стабилизирующие мутации из-за двух присущих предубеждений.Одним из смещений является эффект масштабирования, при котором величина оценки эпистаза зависит от функциональной оценки участвующих вариантов. Другая ошибка — это ошибка выборки, присущая глубокому мутационному сканированию, вызванная его зависимостью от отбора; очень вредные мутации либо не наблюдаются, либо наблюдаются только тогда, когда они сочетаются с полезными мутациями.
Чтобы устранить эти предубеждения, мы разработали третью стратегию, которая использует метрику на основе эпистаза, которую мы назвали потенциацией партнера (рис.3 А ). Партнерская потенциация количественно определяет степень, в которой отдельная одиночная мутация ( a ) улучшает или потенцирует функциональный эффект ее партнерских одиночных мутаций ( b 1 , b 2 ,… b x ) в коллекции двойных мутантов, в которой он обнаружен ( ab 1 , ab 2 ,… ab x ). В данном двойном мутанте ( ab ) a имеет нормализованный по партнеру показатель эпистаза с другой мутацией b ( P a → b ), рассчитанный по формуле (Ур. 5 )
Рис. 3.
Потенцирование партнера выявляет стабилизирующие мутации. ( A ) Потенцирование партнера рассчитывается для запрашиваемой мутации a , которая образует варианты с двойными мутациями с мутациями b 1 , b 2 , b 3 … b n . Нормализованные по партнеру оценки эпистаза рассчитываются для a путем деления оценки эпистаза каждой комбинации двойных мутантов, в которой обнаруживается a , на функциональную оценку мутации партнера (показан пример расчета для b 1 ). ).Потенцирование партнера a рассчитывается как среднее из нормализованных баллов эпистаза (в данном случае баллов b 1 … b n ). ( B ) Потенцирование партнера нанесено на график для каждой отдельной мутации в зависимости от ее функциональной оценки. Известные стабилизирующие (A20R, L30K и D34T) и повышающие активность (K21R и Q35R) мутации выделены красным и синим цветом соответственно. Мутации с баллом потенцирования партнера выше 0.4 и функциональная оценка выше 0,9 считались стабилизирующими. ( C ) Стабилизирующие мутации подтверждены термической денатурацией. Домен WT hYAP65 WW, известный стабилизирующий мутант D34T и четыре кандидатных стабилизирующих мутанта (L30I, I33R, Q35K и T36R) показаны черным, фиолетовым, красным, зеленым, оранжевым и синим цветом соответственно. ( D ) Показаны положения в структуре ЯМР hYAP65 (код 1k9q банка данных белков) со стабилизирующими вариантами, оцененные по потенцированию партнера, окрашенные величиной среднего балла потенцирования партнера с использованием программного обеспечения PyMol.Стабилизирующие мутации распространены по всему домену WW. Выделены позиции ранее известных (верхний индекс 1) и ранее неизвестных (верхний индекс 2) подтвержденных стабилизирующих мутаций (20, 30, 34, 35).
Оценка партнерского потенцирования a ( PP a ) рассчитывается как среднее значение показателей эпистаза, нормализованных по партнеру ( P a → b 1 , P a → b 2 ,… P a → bx ).Мы подсчитали баллы потенцирования партнеров для мутаций, произошедших по крайней мере в 10 двойных мутантах. Потенцирование партнера учитывает функциональные эффекты, передаваемые мутациями партнера, уменьшая влияние как ошибок масштабирования, так и выборки. В отличие от изменения представления или среднего эпистаза, потенцирование партнера отделяет все три известные стабилизирующие мутации от основной массы точек, а также известные мутации, повышающие активность (рис. 3 B ). Как и ожидалось, мутации с высокими показателями потенцирования партнера часто приводили к положительному эпистазу (рис.S3 G ).
Мы определили список кандидатов из 15 стабилизирующих мутаций, имеющих балл потенцирования партнера более 0,4 и функциональный балл более 0,9 (таблица S2). Список содержит три известные стабилизирующие мутации и не включает ни одной из известных мутаций, повышающих активность. Мы химически синтезировали шесть вариантов стабилизирующего домена WW (D10Q, P12H, L30I, Q35K, I33R и T36R), а также известный стабилизирующий вариант D34T в качестве положительного контроля (20), чтобы охарактеризовать их стабильность термической денатурацией (рис.3 С ). Спектроскопию кругового дихроизма в дальнем УФ-диапазоне использовали для записи кривых денатурации, из которых были извлечены данные ΔG сворачивания и ΔΔG сворачивания (таблица S2). Стабильность вариантов D10Q и P12H не могла быть определена количественно из-за отсутствия предпереходных базовых линий в результате их низкой стабильности. Эти сильно дестабилизирующие мутации расположены рядом с N-концом домена WW. Эти мутации могут действовать, чтобы стабилизировать интерфейс фагового капсида-WW домен, и, следовательно, они могут стабилизировать WW домен в фаговом анализе, но не в контексте изолированного WW домена.Из оставшихся пяти вариантов мутации L30I, D34T и Q35K привели к значительной стабилизации, I33R был нейтральной мутацией, а T36R немного дестабилизировал. Среди этих пяти вариантов ΔΔG сворачивание сильно коррелировало с потенциацией партнера, но не со средним эпистазом или функциональной оценкой (ρ Спирмена = -0,81) (рис. S4). Более того, идентификация двух стабилизирующих мутаций в группе с тремя известными стабилизирующими мутациями показывает, что термодинамическая стабильность, фундаментальное свойство белка, неявно присутствует в крупномасштабных функциональных данных.
Чтобы оценить уровень ложноотрицательных результатов нашего подхода, мы сравнили наши результаты с исследованием стабильности в WW-домене Pin1, который имеет высокую степень последовательности и структурной гомологии с WW-доменом hYAP65. Стабильность 47 мутантов по аланину или глицину, распределенных по всему домену Pin1, оценивалась термической денатурацией (23). Из этих мутантов один (2,1%) мутант обладал значительной стабилизацией, что позволяет предположить, что среди 646 возможных одиночных мутаций в домене hYAP65 должно существовать около 14 стабилизирующих мутаций.Предполагая, что наш уровень валидации 33% (2/6) распространяется на все 12 мутаций-кандидатов, мы ожидаем найти в общей сложности четыре стабилизирующие мутации в дополнение к трем известным стабилизирующим мутациям. Эти семь мутаций составляют половину от общего числа, предсказанного на основе данных Pin1. Ложноотрицательные результаты могут быть вызваны неполнотой данных, которые позволили рассчитать баллы потенцирования партнера для 192 из 646 возможных одиночных мутаций, различиями в стабильности между Pin1 и hYAP65 или внутренними ограничениями этого подхода.Мы сравнили нашу скорость проверки (2/6) со скоростью случайного обнаружения стабилизирующих мутаций, предложенной данными Pin1 (1/47), и обнаружили, что наша скорость была значительно выше (точный биномиальный тест, P = 0,0067). Кроме того, мы провели анализ ограниченного набора данных высокого качества и получили почти идентичные результаты (рис. S5).
15 выявленных нами мутаций-кандидатов встречаются всего в восьми положениях. Эти позиции разбросаны по всему домену WW и не ограничиваются областями петель (рис.2 и 3 D ). Фактически, кандидатные стабилизирующие мутации возникают в положениях как в петлях, так и в цепях, а также в положениях, контактирующих и неконтактных с лигандом. Одна мутация, L30I, увеличила T m на поразительную величину 12 ° ° C и, таким образом, является более стабилизирующей, чем любая другая известная стабилизирующая мутация в домене hYAP65 WW. Положение 30 контактирует с пептидным лигандом и является сайтом другой известной стабилизирующей мутации, L30K. Идентификация L30I подчеркивает необходимость поиска стабилизирующих мутаций на основе функциональных данных, поскольку эти мутации не будут препятствовать связыванию пептида, даже если они возникают в месте контакта.
Наконец, мы использовали FoldX (24), широко используемый вычислительный инструмент для прогнозирования термодинамического воздействия мутаций в белках, чтобы проанализировать влияние одиночных мутаций на стабильность домена WW. Ни одна из известных или возможных стабилизирующих мутаций не была классифицирована FoldX (Dataset S1) как стабилизирующая. Этот результат подчеркивает сложность компьютерного предсказания термодинамического воздействия мутаций в белках и подчеркивает эффективность нашей стратегии.
Кандидат, стабилизирующий и активирующий мутации, синергизирует для улучшения функции.
Наши данные дают возможность подробно изучить поведение кандидатов на стабилизирующие мутации в белке. Мы проверили теорию о том, что кандидатные стабилизирующие мутации позволяют приобретать усиливающие активность, но дестабилизирующие мутации (10, 25, 26). Мы классифицировали единичные мутации, которые оказали положительное влияние на функцию, но не были классифицированы как стабилизирующие мутации-кандидаты как активирующие мутации. Среди вариантов с функциональной оценкой выше, чем WT, те варианты, которые содержат две активирующие мутации, обычно имеют более высокие функциональные оценки, чем варианты с одной активирующей мутацией (и не имеют стабилизирующей мутации кандидата) (рис.4 А ). Варианты со стабилизирующими мутациями-кандидатами (и без активирующих мутаций) имели более высокие функциональные баллы, чем варианты, которые полагались исключительно на активирующие мутации, что позволяет предположить, что домен WW является лишь незначительно стабильным (рис.4 A ) (критерий суммы рангов Вилкоксона, P = 6,65 × 10 −14 ). Наибольшее функциональное увеличение баллов произошло у двойных мутантов, которые сочетают кандидатную стабилизирующую мутацию с активирующей мутацией (критерий суммы рангов Вилкоксона, P = 8.56 × 10 −13 ) (рис.4 A ).
Рис. 4.
Стабилизирующие мутации в сочетании с активирующими мутациями приводят к большим функциональным улучшениям. ( A ) Мутации классифицировались как стабилизирующие (функциональная оценка> 0,9 и потенцирование партнера> 0,4) или активирующие (функциональная оценка> 1 и потенцирование партнера ≤ 0,4). Двойные мутанты с функциональными баллами выше, чем WT, были сгруппированы в те мутанты, которые несут как активирующие, так и стабилизирующие мутации (фиолетовый; n = 170), единственную стабилизирующую мутацию, но не активирующие мутации (красный; n = 156), единственную активирующую мутацию. мутация, но без стабилизирующей мутации (сплошной синий; n = 1071) или две активирующие мутации (синий пунктир; n = 852).Представлены функциональные распределения баллов для каждого класса. Кривые для вариантов с более высокими функциональными баллами, чем WT, содержащими парные стабилизирующие мутации, не строятся из-за их низкого числа ( n = 4). ( B ) Для каждой стабилизирующей (красный) и активирующей мутации (синий) была рассчитана доля спасенных вредных одиночных мутаций (т.е. обнаруженных у двойных мутантов с более высокими функциональными баллами, чем WT). Анализ был ограничен вредными мутациями, с которыми были связаны как стабилизирующие, так и активирующие мутации в двойных мутантах в нашей библиотеке.Экспериментально подтвержденные полезные мутации выделены жирным шрифтом; 1 обозначает стабилизирующие мутации, подтвержденные в этом исследовании, 2 обозначает ранее идентифицированные стабилизирующие мутации, а 3 обозначает ранее идентифицированные активирующие мутации.
Мы сравнили способность кандидатов стабилизировать и активировать мутации для спасения вредных мутаций. Стабилизирующие мутации должны более эффективно спасать вредные мутации, потому что большинство вредных мутаций являются дестабилизирующими, а дефект решается напрямую путем стабилизации мутаций, но не активации мутаций.Чтобы проверить это предсказание, мы определили события «спасения», в которых единичная вредоносная мутация сочеталась с кандидатом, стабилизирующим или активирующим мутацию, у двойного мутанта с функциональной оценкой не ниже, чем у WT. Чтобы избежать ошибок выборки, мы ограничили этот анализ набором вредных мутаций в сочетании с активирующими и стабилизирующими мутациями-кандидатами в функциональных данных двойных мутантов. В этом наборе вредные мутации, которые устраняются стабилизирующими мутациями-кандидатами и активирующими мутациями, в значительной степени перекрываются (~ 70%).Однако стабилизирующие мутации-кандидаты спасали в среднем в три раза больше вредных мутаций, чем активирующие мутации. Чтобы изучить эффекты спасения для каждой мутации, мы ранжировали мутации, стабилизирующие и активирующие кандидатов, по доле вредных мутаций, которые они спасли. Независимо проверенные стабилизирующие и активирующие мутации спасли самую большую часть вредных мутаций в своем классе (рис. 4 B ). Таким образом, кандидатные стабилизирующие и активирующие мутации могут спасти многие из одних и тех же вредных мутаций, но кандидатные стабилизирующие мутации повышают толерантность к вредным мутациям в гораздо большей степени, чем активирующие мутации.
Стабилизирующие мутации спасают дестабилизирующие мутации за счет буферизации снижения стабильности, тогда как активирующие мутации оказывают свое спасательное действие, буферизуя функциональные затраты за счет повышения активности. Доля мутаций, спасенных данной активирующей или стабилизирующей мутацией-кандидатом, в большей степени коррелировала с функциональной оценкой активирующих мутаций (ρ = 0,92 Спирмена), чем мутации-кандидаты стабилизирующей мутации (ρ = 0,59; P ≤ 1 × 10 −4 ). ) (Рис.S6). Это открытие предполагает, что спасение путем активации мутаций происходит за счет обмена стоимостью, а спасение с помощью стабилизирующих мутаций-кандидатов происходит с помощью другого механизма.
Обсуждение
Подходы с высокой пропускной способностью, такие как глубокое мутационное сканирование, могут измерять функцию вариантов белка в беспрецедентном масштабе. В виде простого списка крупномасштабные функциональные данные, полученные с помощью этих подходов, идентифицируют полезные и вредные мутации, а также позиции, важные для активности белка.Здесь мы показали, что можем использовать эти данные, чтобы идентифицировать признак, который не сразу очевиден только по функциональным оценкам: идентичность стабилизирующих мутаций. Для достижения этой цели мы разработали метрику потенцирования партнера, которая позволила нам идентифицировать стабилизирующие мутации без необходимости явно измерять стабильность. Мы нашли 15 кандидатных стабилизирующих мутаций и подтвердили 2 ранее неизвестные мутации среди ~ 600 возможных одиночных мутаций в домене WW, что подтверждает мнение о том, что стабилизирующие мутации — редкость.Три из этих мутаций были ранее идентифицированы с помощью рационального дизайна, но большинство мутаций нельзя было предсказать. Таким образом, можно проанализировать крупномасштабные функциональные данные, чтобы выявить по крайней мере одно фундаментальное свойство белка.
Кроме того, мы охарактеризовали эпистаз в домене WW в массовом масштабе. Большинство одиночных мутаций не проявляли сильного эпистаза при сочетании, и не было средней тенденции к положительному или отрицательному эпистазу. Недавно крупномасштабный анализ эпистаза в протеазе ВИЧ описал географическое обогащение эпистаза (27).В данных WW мы нашли аналогичные доказательства сильных эпистатических взаимодействий, происходящих между отдельными регионами. Кроме того, наши данные показывают, что возникновение положительных и отрицательных эпистатических взаимодействий коррелировано в позициях в домене WW, выделяя ограниченное количество горячих точек, где эпистатические взаимодействия, как положительные, так и отрицательные, наиболее вероятны.
Наш анализ позволил нам классифицировать большое количество мутаций как потенциально активирующие или стабилизирующие.Взаимодействие между активирующими и стабилизирующими мутациями имеет значение как для эволюции белков, так и для белковой инженерии (9, 10, 16). Эта работа предлагает явную крупномасштабную проверку теории эволюции белков, которая предсказывает, что стабилизирующие мутации допускают существование других активирующих, но дестабилизирующих мутаций. Обнаружение того, что двойные мутанты WW-домена с одной стабилизирующей мутацией-кандидатом и одной вредоносной мутацией обладают повышенной функцией по сравнению с мутантами с одной активирующей мутацией и одной вредоносной мутацией, подтверждает эту теорию.Кроме того, мы обнаружили, что кандидатные стабилизирующие мутации повышают толерантность к вредным мутациям в большей степени, чем активирующие мутации. Этот результат иллюстрирует важность стабилизации мутаций для сохранения разнообразия во время эволюции белка.
Вычислительные и экспериментальные подходы были разработаны для идентификации стабилизирующих мутаций. Вычислительные методы обычно основываются на физико-химических моделях для оценки термодинамического воздействия мутаций (28, 29). Стабилизирующие мутации также можно идентифицировать путем анализа эволюционной консервации белков гипертермофильных организмов (30, 31).Рациональный дизайн основан на структуре белка, а также на знаниях экспериментатора для предсказания стабилизирующих мутаций (32). Методы, основанные на отборе, включая направленную эволюцию, пытаются различать стабилизирующие мутации путем отбора по активности среди библиотеки вариантов белка в условиях, которые включают высокую температуру, денатурант или присутствие протеазы (31–33). Уровень валидации описанного здесь подхода (33%) в целом аналогичен другим подходам; однако стабилизирующие, но дезактивирующие мутации, мешающие другим стратегиям, устраняются.
Систематические анализы, представленные здесь, могут позволить нам распутать и, следовательно, количественно оценить другие свойства, которые влияют на функцию белка. Например, для предсказания структуры белка могут быть полезны крупномасштабные функциональные данные белка, которые выявляют предпочтения аминокислот в определенных структурных элементах (например, нехватку остатков пролина в β-цепях) и функциональные эффекты мутаций, которые происходят в пространственно проксимальных позиции. Осуществимость этого подхода иллюстрируется существующими методами структурного прогнозирования, которые основаны на этих концепциях, но требуют обширных существующих данных о выравнивании последовательностей или структурных обучающих данных (34, 35).Другой пример относится к пониманию механизма фермента, который может быть раскрыт путем анализа паттерна мутаций, которые увеличивают или уменьшают каталитическую активность в крупномасштабных функциональных данных белка. В частности, изучение редких сильно активирующих мутаций представляет собой систематический метод изучения механизма. Наконец, интерфейсы взаимодействия белок-белок могут быть детально картированы путем анализа крупномасштабных функциональных данных белка, собранных в присутствии и в отсутствие взаимодействующего белкового партнера.Зависимые от партнера изменения в функции варианта будут указывать на положения, важные для взаимодействия связывания, и предпочтение аминокислот в этих положениях может выявить природу поверхности связывания. Таким образом, мы прогнозируем, что растущая доступность крупномасштабных функциональных данных о белках предоставит новые захватывающие инструменты для понимания того, как функционируют белки.
Материалы и методы
Мы кратко обсуждаем здесь ключевые методы и отсылаем читателей к SI Text для получения полных экспериментальных и аналитических деталей.
Создание библиотеки фагового дисплея, отбора и секвенирования домена WW.
Мы выполнили фаговый дисплей и селекцию, как описано ранее (2). Было проведено три раунда отбора библиотеки WW-домена против пептида GTPPPPYTVG, связанного с магнитными шариками. Библиотеки высокопроизводительного секвенирования получали с помощью ПЦР, а затем секвенировали на GAIIx (Illumina).
Высокопроизводительное секвенирование и качественная фильтрация.
Переменная область из 102 оснований была секвенирована с использованием частично перекрывающихся считываний для повышения качества (2).Данные анализировали с помощью пакета программ Enrich (36).
Расчет вариантов функциональных баллов.
Для расчета функциональных баллов по вариантам мы использовали данные с поправкой на неспецифический перенос из последовательных раундов отбора, чтобы построить линейную модель для каждого варианта. Наклон линии в этой модели пропорционален функции варианта; варианты, которые обогащают на протяжении всего отбора, имеют положительные наклоны, тогда как варианты, которые истощаются, имеют отрицательные наклоны. Фильтры качества подгонки ( R 2 > 0.75) использовались, как описано в SI Text .
Благодарности
Мы благодарим Чарли Ли и Джея Шендуре за помощь в секвенировании ДНК, а также Элханана Боренштейна, Кристин Куич, Дэвида Бейкера, Ричарда Маклафлина и Карлоса Арайю Родригеса за полезные комментарии и обсуждение. С.Ф. является исследователем Медицинского института Говарда Хьюза. Эта работа была поддержана грантами Национального института здравоохранения F32GM084699 (для D.M.F), GM051105 (для J.W.K.) и P41GM103533 (для S.F.).
Сноски
-
Авторы: C.L.A., D.M.F. и S.F. спланированное исследование; C.L.A., D.M.F., W.C. и I.M. проводили исследования; C.L.A., D.M.F., W.C., J.W.K. и S.F. проанализированные данные; и C.L.A., D.M.F., J.W.K. и S.F. написал газету.
-
Авторы заявляют об отсутствии конфликта интересов.
-
Эта статья представляет собой прямое представление PNAS.
-
Размещение данных: необработанные данные последовательности, представленные в этой статье, были депонированы в архиве чтения последовательностей (номер доступа.SRA058752).
-
Эта статья содержит вспомогательную информацию на сайте www.pnas.org/lookup/suppl/doi:10.1073/pnas.1209751109/-/DCSupplemental.
Структурные и функциональные свойства шипового белка SARS-CoV-2: разработка потенциального антивирусного препарата для COVID-19
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. . Новый коронавирус от пациентов с пневмонией в Китае, 2019. N Engl J Med. 2020; 382: 727–33.
PubMed
PubMed Central
CAS
Google Scholar
Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, et al. Анализ терапевтических целей для SARS-CoV-2 и открытие потенциальных лекарств с помощью вычислительных методов. Acta Pharm Sin B. 2020; 10: 766–88.
PubMed
PubMed Central
Google Scholar
Лю Ц., Чжоу К., Ли И, Гарнер Л.В., Уоткинс С.П., Картер Л.Дж. и др. Исследования и разработки терапевтических агентов и вакцин против COVID-19 и связанных с ним заболеваний, связанных с коронавирусом человека.ACS Cent Sci. 2020; 6: 315–31.
PubMed
PubMed Central
CAS
Google Scholar
Сандерс Дж. М., Моног М. Л., Йодловски Т. З., Катрелл Дж. Б.. Фармакологические методы лечения коронавирусной болезни 2019 (COVID-19): обзор. ДЖАМА. 2020; 323: 1824–36.
CAS
Google Scholar
Лу Р, Чжао Х, Ли Дж, Ниу П, Ян Б., Ву Х и др. Геномная характеристика и эпидемиология нового коронавируса 2019 г .: влияние на происхождение вируса и связывание с рецептором.Ланцет. 2020; 395: 565–74.
PubMed
PubMed Central
CAS
Google Scholar
Chen L, Liu W., Zhang Q, Xu K, Ye G, Wu W. и др. Подход mNGS на основе РНК позволяет идентифицировать новый коронавирус человека из двух отдельных случаев пневмонии во время вспышки в Ухане в 2019 году. Emerg Microbes Infect. 2020; 9: 313–9.
PubMed
PubMed Central
CAS
Google Scholar
Чан Дж. Ф., Кок К. Х., Чжу З., Чу Х, То К. К., Юань С. и др.Геномная характеристика нового патогенного для человека коронавируса 2019 года, выделенного от пациента с атипичной пневмонией после посещения Ухани. Emerg Microbes Infect. 2020; 9: 221–36.
PubMed
PubMed Central
CAS
Google Scholar
Летко М., Марзи А., Мюнстер В. Функциональная оценка входа в клетки и использования рецепторов для SARS-CoV-2 и других бета-коронавирусов линии B. Nat Microbiol. 2020; 5: 562–9.
CAS
Google Scholar
Фер А.Р., Перлман С. Коронавирусы: обзор их репликации и патогенеза. Методы Мол биол. 2015; 1282: 1–23.
PubMed
PubMed Central
CAS
Google Scholar
Морс Дж. С., Лалонд Т., Сюй С., Лю В. Р.. Уроки прошлого: возможные варианты срочной профилактики и лечения тяжелых острых респираторных инфекций, вызванных 2019-nCoV. Chembiochem. 2020; 21: 730–8.
PubMed
PubMed Central
CAS
Google Scholar
Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. Белок-спайк коронавируса — это гибридный белок вируса класса I: структурная и функциональная характеристика комплекса слитого ядра. J Virol. 2003; 77: 8801–11.
PubMed
PubMed Central
CAS
Google Scholar
Watanabe Y, Allen JD, Wrapp D, McLellan JS, Crispin M. Сайт-специфический гликановый анализ шипа SARS-CoV-2. Наука. 2020; 369: 330–3.
PubMed
PubMed Central
CAS
Google Scholar
Xia S, Zhu Y, Liu M, Lan Q, Xu W., Wu Y, et al. Механизм слияния 2019-nCoV и ингибиторов слияния, нацеленных на домен HR1 в спайковом белке. Cell Mol Immunol. 2020; 17: 765–7.
PubMed
CAS
Google Scholar
Tang T, Bidon M, Jaimes JA, Whittaker GR, Daniel S. Механизм слияния мембран коронавируса представляет собой потенциальную мишень для противовирусных разработок. Antivir Res. 2020; 178: 104792.
PubMed
CAS
Google Scholar
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O и др. Крио-ЭМ структура спайка 2019-нКоВ в конформации до слияния. Наука. 2020; 367: 1260–3.
PubMed
PubMed Central
CAS
Google Scholar
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Структура, функция и антигенность гликопротеина шипа SARS-CoV-2. Клетка. 2020; 181: 281–92 e286.
PubMed
PubMed Central
CAS
Google Scholar
Бертрам С., Дейкман Р., Хабьян М., Хойрих А., Гирер С., Гловакка И. и др. TMPRSS2 активирует человеческий коронавирус 229E для катепсин-независимого входа в клетки-хозяева и экспрессируется в вирусных клетках-мишенях в респираторном эпителии. J Virol. 2013; 87: 6150–60.
PubMed
PubMed Central
CAS
Google Scholar
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T., Erichsen S, et al. Вход в клетки SARS-CoV-2 зависит от ACE2 и TMPRSS2 и блокируется клинически доказанным ингибитором протеазы.Клетка. 2020; 181: 271–80.e8.
PubMed
PubMed Central
CAS
Google Scholar
Du L, Kao RY, Zhou Y, He Y, Zhao G, Wong C, et al. Расщепление спайкового белка коронавируса SARS протеазным фактором Ха связано с вирусной инфекционностью. Biochem Biophys Res Commun. 2007; 359: 174–9.
PubMed
PubMed Central
CAS
Google Scholar
Ван Ц., Чжан И, Ву Л., Ню С., Сонг С., Чжан З. и др.Структурная и функциональная основа проникновения SARS-CoV-2 с использованием человеческого ACE2. Клетка. 2020; 181: 894–904.e9.
PubMed
PubMed Central
CAS
Google Scholar
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S и др. Структура спайкового домена, связывающего рецептор SARS-CoV-2, связанного с рецептором ACE2. Природа. 2020; 581: 215–20.
PubMed
CAS
Google Scholar
Xia S, Yan L, Xu W., Agrawal AS, Algaissi A, Tseng CK, et al. Ингибитор слияния пан-коронавируса, нацеленный на домен HR1 спайка коронавируса человека. Sci Adv. 2019; 5: eaav4580.
PubMed
PubMed Central
CAS
Google Scholar
Миллет Дж. К., Уиттакер ГР. Физиологические и молекулярные триггеры слияния мембран SARS-CoV и проникновения в клетки-хозяева. Вирусология. 2018; 517: 3–8.
PubMed
CAS
Google Scholar
Chambers P, Pringle CR, Easton AJ. Последовательности гептадных повторов расположены рядом с гидрофобными участками в нескольких типах гликопротеинов слияния вирусов. J Gen Virol. 1990; 71: 3075–80.
PubMed
CAS
Google Scholar
Робсон Б. Компьютеры и вирусные болезни. Предварительные биоинформатические исследования по разработке синтетической вакцины и профилактического пептидомиметического антагониста против коронавируса SARS-CoV-2 (2019-nCoV, COVID-19).Comput Biol Med. 2020; 119: 103670.
PubMed
PubMed Central
CAS
Google Scholar
Xia S, Xu W., Wang Q, Wang C, Hua C, Li W, et al. Ингибиторы слияния мембран на основе пептидов, нацеленные на домены HR1 и HR2 шипового белка HCoV-229E. Int J Mol Sci. 2018; 19: 487. https://doi.org/10.3390/ijms187.
Артикул
PubMed Central
CAS
Google Scholar
Лу Г, Ван Кью, Гао ГФ. От летучей мыши к человеку: особенности всплеска, определяющие «скачок от хозяина» коронавирусов SARS-CoV, MERS-CoV и других. Trends Microbiol. 2015; 23: 468–78.
PubMed
PubMed Central
CAS
Google Scholar
Лю С., Сяо Дж., Чен И, Хе И, Ниу Дж., Эскаланте С.Р. и др. Взаимодействие между участками 1 и 2 гептадных повторов в спайковом белке коронавируса, связанного с SARS: влияние на механизм слияния вируса и идентификация ингибиторов слияния.Ланцет. 2004; 363: 938–47.
PubMed
PubMed Central
CAS
Google Scholar
Yu Y, Deng YQ, Zou P, Wang Q, Dai Y, Yu F и др. Вирусный инактиватор на основе пептидов подавляет вирусную инфекцию Зика у беременных мышей и плодов. Nat Commun. 2017; 8: 15672.
PubMed
PubMed Central
CAS
Google Scholar
Weissenhorn W., Dessen A, Calder LJ, Harrison SC, Skehel JJ, Wiley DC.Структурная основа слияния мембран вирусами в оболочке. Mol Membr Biol. 1999; 16: 3–9.
PubMed
CAS
Google Scholar
Gui M, Song W, Zhou H, Xu J, Chen S, Xiang Y, et al. Структуры криоэлектронной микроскопии гликопротеина шипа SARS-CoV выявляют предварительное конформационное состояние для связывания рецептора. Cell Res. 2017; 27: 119–29.
PubMed
CAS
Google Scholar
Hulswit RJ, де Хаан, Калифорния, Bosch BJ. Пиковый белок коронавируса и изменения тропизма. Adv Virus Res. 2016; 96: 29–57.
PubMed
PubMed Central
CAS
Google Scholar
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Структурная основа для распознавания SARS-CoV-2 полноразмерным человеческим ACE2. Наука. 2020; 367: 1444–8.
PubMed
PubMed Central
CAS
Google Scholar
Цуй Дж., Ли Ф., Ши З.Л. Происхождение и эволюция патогенных коронавирусов. Nat Rev Microbiol. 2019; 17: 181–92.
PubMed
CAS
Google Scholar
Донохью М., Сие Ф., Баронас Э., Годбаут К., Госселин М., Стальяно Н. и др. Новая карбоксипептидаза, связанная с ангиотензинпревращающим ферментом (ACE2), превращает ангиотензин I в ангиотензин 1-9. Circ Res. 2000; 87: E1–9.
PubMed
PubMed Central
CAS
Google Scholar
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Ангиотензин-превращающий фермент 2 (ACE2) как рецептор SARS-CoV-2: молекулярные механизмы и потенциальная терапевтическая мишень. Intensive Care Med. 2020; 46: 586–90.
PubMed
CAS
Google Scholar
Шан Дж, Ван И, Ло Ц., Йе Г, Гэн К., Ауэрбах А. и др. Механизмы входа в клетки SARS-CoV-2. Proc Natl Acad Sci USA. 2020; 117: 11727–34.
PubMed
CAS
Google Scholar
Чен И, Го И, Пань И, Чжао ЗДж. Анализ структуры рецепторного связывания 2019-nCoV. Biochem Biophys Res Commun. 2020; 525: 135–40.
PubMed Central
Google Scholar
Ван И, Шан Дж., Грэм Р., Барик Р.С., Ли Ф. Распознавание рецепторов новым коронавирусом из Ухани: анализ, основанный на десятилетних структурных исследованиях коронавируса SARS. J Virol. 2020; 94: e00127–20.
PubMed
PubMed Central
Google Scholar
Tortorici MA, Walls AC, Lang Y, Wang C, Li Z, Koerhuis D, et al. Структурная основа прикрепления коронавируса человека к рецепторам сиаловой кислоты. Nat Struct Mol Biol. 2019; 26: 481–9.
PubMed
PubMed Central
Google Scholar
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. Спайковый гликопротеин нового коронавируса 2019-nCoV содержит фурин-подобный сайт расщепления, отсутствующий в CoV той же клады.Antivir Res. 2020; 176: 104742.
PubMed
CAS
Google Scholar
Рабаан А.А., Аль-Ахмед С.Х., Хак С., Сах Р., Тивари Р., Малик Ю.С. и др. SARS-CoV-2, SARS-CoV и MERS-COV: сравнительный обзор. Infez Med. 2020; 28: 174–84.
PubMed
CAS
Google Scholar
Хасан А., Парай Б.А., Хуссейн А., Кадир Ф.А., Аттар Ф., Азиз Ф.М. и др. Обзор примирования расщепления белка-шипа на коронавирусе ангиотензин-превращающим ферментом-2 и фурином.J Biomol Struct Dyn. 2020; 22: 1–9.
Google Scholar
Миллет Дж. К., Уиттакер ГР. Протеазы клеток-хозяев: критические детерминанты тропизма и патогенеза коронавируса. Virus Res. 2015; 202: 120–34.
PubMed
CAS
Google Scholar
Claas EC, Osterhaus AD, van Beek R, De Jong JC, Rimmelzwaan GF, Senne DA, et al. Человеческий грипп Вирус H5N1, относящийся к высокопатогенному вирусу птичьего гриппа.Ланцет. 1998; 351: 472–7.
PubMed
CAS
Google Scholar
Kido H, Okumura Y, Takahashi E, Pan HY, Wang S, Yao D, et al. Роль протеаз клеток хозяина в патогенезе гриппа и полиорганной недостаточности. Biochim Biophys Acta. 2012; 1824: 186–94.
PubMed
CAS
Google Scholar
Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S.TMPRSS2 и ADAM17 по-разному расщепляют ACE2, и только протеолиз с помощью TMPRSS2 увеличивает вход, вызванный спайк-белком коронавируса тяжелого острого респираторного синдрома. J Virol. 2014; 88: 1293–307.
PubMed
PubMed Central
Google Scholar
Limburg H, Harbig A, Bestle D, Stein DA, Moulton HM, Jaeger J, et al. TMPRSS2 является основной активирующей протеазой вируса гриппа A в первичных клетках дыхательных путей человека и вируса гриппа B в пневмоцитах человека II типа.J Virol. 2019; 93: e00649–19.
PubMed
PubMed Central
Google Scholar
Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Характеристика спайкового гликопротеина SARS-CoV-2 при проникновении вируса и его иммунная перекрестная реактивность с SARS-CoV. Nat Commun. 2020; 11: 1620.
PubMed
PubMed Central
CAS
Google Scholar
Кавасэ М, Катаока М, Сирато К., Мацуяма С.Биохимический анализ конформационных промежуточных продуктов гликопротеина шипа коронавируса во время слияния мембран. J Virol. 2019; 93: e00785–19.
PubMed
PubMed Central
CAS
Google Scholar
Harrison SC. Слияние вирусных мембран. Вирусология. 2015; 479-480: 498-507.
PubMed
CAS
Google Scholar
Экерт Д.М., Ким П.С. Механизмы слияния вирусных мембран и его ингибирование.Анну Рев Биохим. 2001; 70: 777–810.
PubMed
CAS
Google Scholar
Дхама К., Шарун К., Тивари Р., Дадар М., Малик Ю.С., Сингх К.П. и др. COVID-19, новая коронавирусная инфекция: достижения и перспективы в разработке и разработке вакцин, иммунотерапевтических и терапевтических средств. Hum Vaccin Immunother. 2020; 16: 1232–8
PubMed
CAS
Google Scholar
Чжэн М., Сонг Л. Новые эпитопы антител доминируют в антигенности спайкового гликопротеина у SARS-CoV-2 по сравнению с SARS-CoV. Cell Mol Immunol. 2020; 17: 536–8.
PubMed
CAS
Google Scholar
Гао Кью, Бао Л., Мао Х, Ван Л., Сюй К., Ян М. и др. Быстрая разработка инактивированной вакцины-кандидата от SARS-CoV-2. Наука. 2020; 369: 77–81.
PubMed
CAS
Google Scholar
Coleman CM, Liu YV, Mu H, Taylor JK, Massare M, Flyer DC, et al. Очищенные наночастицы белка-шипа коронавируса индуцируют у мышей нейтрализующие антитела к коронавирусу. Вакцина. 2014; 32: 3169–74.
PubMed
PubMed Central
CAS
Google Scholar
Тиан Х, Ли К., Хуанг А., Ся С., Лу С., Ши Зи и др. Сильное связывание нового шипового белка коронавируса 2019 года человеческими моноклональными антителами, специфичными для коронавируса SARS. Emerg Microbes Infect.2020; 9: 382–5.
PubMed
PubMed Central
CAS
Google Scholar
Ван С., Ли В., Драбек Д., Окба НМА, ван Хаперен Р., Остерхаус А. и др. Человеческое моноклональное антитело, блокирующее инфекцию SARS-CoV-2. Nat Commun. 2020; 11: 2251.
PubMed
PubMed Central
CAS
Google Scholar
Chen XY, Li R, Pan ZW, Qian CF, Yang Y, You RR, et al. Человеческие моноклональные антитела блокируют связывание спайкового белка SARS-CoV-2 с рецептором ангиотензинпревращающего фермента 2.Cell Mol Immunol. 2020; 17: 647–9.
PubMed
CAS
Google Scholar
Wu Y, Li C, Xia S, Tian X, Wang Z, Kong Y, et al. Идентификация полностью человеческих однодоменных антител против SARS-CoV-2. Клеточный микроб-хозяин. 2020; 27: 891–8.e5. https://doi.org/10.1101/2020.03.30.015990.
Артикул
PubMed
PubMed Central
CAS
Google Scholar
Pinto D, Park Y-J, Beltramello M, Walls AC, Tortorici MA, Bianchi S, et al. Структурный и функциональный анализ мощного нейтрализующего антитела к сарбековирусу. bioRxiv. 2020; 2020.04.07.023903. https://doi.org/10.1101/2020.04.07.023903.
Ju B, Zhang Q, Ge X, Wang R, Yu J, Shan S, et al. Сильные нейтрализующие антитела человека, вызванные инфекцией SARS-CoV-2. bioRxiv. 2020; https://doi.org/10.1101/2020.03.21.9
Chi X, Yan R, Zhang J, Zhang G, Zhang Y, Hao M, et al.Мощное нейтрализующее человеческое антитело обнаруживает, что N-концевой домен белка Spike SARS-CoV-2 является уязвимым местом. bioRxiv. 2020. https://doi.org/10.1101/2020.05.08.083964.
Xia S, Liu MQ, Wang C, Xu W., Lan QS, Feng SL, et al. Ингибирование инфекции SARS-CoV-2 (ранее 2019-nCoV) с помощью мощного ингибитора слияния панкоронавируса, нацеленного на его спайковый белок, обладающий высокой способностью опосредовать слияние мембран. Cell Res. 2020; 30: 343–55.
PubMed
CAS
Google Scholar
Zhu Y, Yu D, Yan H, Chong H, He Y. Разработка мощных ингибиторов слияния мембран против SARS-CoV-2, нового коронавируса с высокой фузогенной активностью. J Virol. 2020; 94: e00635–20.
PubMed
PubMed Central
CAS
Google Scholar
Musarrat F, Chouljenko V, Dahal A, Nabi R, Chouljenko T, Jois SD, et al. Препарат против ВИЧ Нелфинавир Мезилат (Вирасепт) является мощным ингибитором слияния клеток, вызванного гликопротеином SARS-CoV-2 Spike (S), что требует дальнейшей оценки в качестве противовирусного средства против инфекций COVID-19.J Med Virol. 2020; 10.1002 / jmv.25985.
Uno Y. Терапия мезилатом Camostat для COVID-19. Intern Emerg Med. 2020; 1–2. https://doi.org/10.1007/s11739-020-02345-9. [Epub перед печатью].
de Wilde AH, Falzarano D, Zevenhoven-Dobbe JC, Beugeling C, Fett C, Martellaro C, et al. Алиспоривир подавляет репликацию коронавирусов MERS и SARS в культуре клеток, но не подавляет заражение коронавирусом SARS на мышиной модели. Virus Res. 2017; 228: 7–13.
PubMed
Google Scholar
Хуанг IC, Bosch BJ, Li WH, Farzan M, Rottier PM. Choe H. SARS-CoV, но не HCoV-NL63, использует катепсины для заражения клеток — проникновение вируса. Нидовирусы: к борьбе с Sars и другими нидовирусами. Болезни. 2006; 581: 335–8.
CAS
Google Scholar
Zhou N, Pan T, Zhang JS, Li QW, Zhang X, Bai C, et al. Гликопептидные антибиотики сильно ингибируют катепсин L в поздних эндосомах / лизосомах и блокируют проникновение вируса Эбола, коронавируса ближневосточного респираторного синдрома (БВРС-КоВ) и коронавируса тяжелого острого респираторного синдрома (ТОРС-КоВ).J Biol Chem. 2016; 291: 9218–32.
PubMed
PubMed Central
CAS
Google Scholar
Нельсон Э.А., Дьялл Дж., Хоэнен Т., Барнс А.Б., Чжоу Х., Лян Дж.Й. и др. Апилимод, ингибитор фосфатидилинозитол-3-фосфат-5-киназы, блокирует проникновение филовирусов и инфицирование. PLoS Negl Trop Dis. 2017; 11: e0005540.
PubMed
PubMed Central
Google Scholar
Hou JZ, Xi ZQ, Niu J, Li W, Wang X, Liang C и др.Ингибирование PIKfyve с помощью YM201636 подавляет рост рака печени за счет индукции аутофагии. Онкол Реп. 2019; 41: 1971–9.
PubMed
CAS
Google Scholar
Сакурай Ю., Колокольцов А.А., Чен С.К., Тидвелл М.В., Баута В.Е., Клугбауэр Н. и др. Двухпористые каналы контролируют проникновение вируса Эбола в клетки-хозяева и являются мишенями для лечения заболеваний. Наука. 2015; 347: 995–8.
PubMed
PubMed Central
CAS
Google Scholar
Артенштейн А.В., Опал СМ. Конверсии пропротеина в здоровье и болезни. N Engl J Med. 2011; 365: 2507–18.
PubMed
CAS
Google Scholar
Хуанг Ц., Ван И, Ли Х, Рен Л., Чжао Дж, Ху И и др. Клинические особенности пациентов, инфицированных новым коронавирусом 2019 г., в Ухане, Китай. Ланцет. 2020; 395: 497–506.
PubMed
PubMed Central
CAS
Google Scholar
Yakala GK, Cabrera-Fuentes HA, Crespo-Avilan GE, Rattanasopa C, Burlacu A, George BL, et al. Ингибирование ФУРИНА снижает ремоделирование сосудов и прогрессирование атеросклеротических поражений у мышей. Артериосклер Thromb Vasc Biol. 2019; 39: 387–401.
PubMed
PubMed Central
CAS
Google Scholar
Zhou M, Zhang Y, Wei H, He J, Wang D, Chen B и др. Ингибитор фурина D6R подавляет эпителиально-мезенхимальный переход в клетках SW1990 и PaTu8988 через сигнальный путь Hippo-YAP.Oncol Lett. 2018; 15: 3192–6.
PubMed
Google Scholar
Leblond J, Laprise MH, Gaudreau S, Grondin F, Kisiel W., Dubois CM. Ингибитор серпиновой протеиназы 8: эндогенный ингибитор фурина, высвобождаемый из тромбоцитов человека. Thromb Haemost. 2006; 95: 243–52.
PubMed
CAS
Google Scholar
Лу И, Хардес К., Дамс С.О., Боттчер-Фрибертсхаузер Э., Штайнметцер Т., Тхан М.Э. и др.Пептидомиметический ингибитор фурина MI-701 в сочетании с осельтамивиром и рибавирином эффективно блокирует распространение высокопатогенных вирусов птичьего гриппа и задерживает высокий уровень устойчивости к осельтамивиру в клетках MDCK. Antivir Res. 2015; 120: 89–100.
PubMed
CAS
Google Scholar
Abidin AZ, DSouza AM, Nagarajan MB, Wang L, Qiu X, Schifitto G, et al. Изменение топологии сети мозга при ВИЧ-ассоциированном нейрокогнитивном расстройстве: новая перспектива функциональной связи.Neuroimage Clin. 2018; 17: 768–77.
PubMed
Google Scholar
Ван Ц., Лю Ц., Чен Ц., Хуанг Х, Сюй М., Хе Т. и др. Установление эталонной последовательности для SARS-CoV-2 и вариационный анализ. J Med Virol. 2020; 92: 667–74.
PubMed
PubMed Central
CAS
Google Scholar
Бесерра-Флорес М., Кардозо Т. Мутация вирусного шипа SARS-CoV-2 G614 демонстрирует более высокий уровень летальности.Int J Clin Pract. 2020; 6: e13525.
Google Scholar
Zhou GY, Zhao Q. Перспективы терапевтических нейтрализующих антител против нового коронавируса SARS-CoV-2. Int J Biol Sci. 2020; 16: 1718–23.
PubMed
PubMed Central
Google Scholar
Wang ML, Cao RY, Zhang LK, Yang XL, Liu J, Xu MY, et al. Ремдесивир и хлорохин эффективно подавляют недавно появившийся новый коронавирус (2019-nCoV) in vitro.Cell Res. 2020; 30: 269–71.
PubMed
PubMed Central
CAS
Google Scholar
Сравнение последовательностей белков на основе физико-химических свойств и энергетической матрицы положения-характеристики
Чжао, Ю., Ли, X. и Ци, З. Новое двухмерное графическое представление последовательности белка и его применение. J. Fiber Bioengineering and Informatics 7 , 23–33 (2014).
Google Scholar
Хуанг Д.& Yu, H. Нормализованные векторы признаков: новый метод сравнения последовательностей без выравнивания, основанный на количестве соседних аминокислот. IEEE / ACM Trans. Comput. Биол. 10 , 457–467 (2013).
Google Scholar
Gotoh, O. Улучшенный алгоритм сопоставления биологических последовательностей. J. Mol. Биол. 162 , 705–708 (1982).
CAS
Google Scholar
Чакраборти, А.& Bandyopadhyay, S. FOGSAA: Алгоритм быстрого оптимального глобального выравнивания последовательностей. Sci. Репутация . 3 , 1746 (2013).
ADS
PubMed
PubMed Central
Google Scholar
Фен, Д. и Дулиттл, Р. Ф. Прогрессивное выравнивание последовательностей как предпосылка для исправления филогенетических деревьев. J. Mol. Evol. 25 , 351–360 (1987).
ADS
CAS
Google Scholar
Брэдли Р.K. et al. Быстрое статистическое выравнивание. PLoS Comput. Биол. 5 , e1000392 (2009).
MathSciNet
PubMed
PubMed Central
Google Scholar
Райнерт, Г., Чу, Д., Сан, Ф. и Уотерман, М. С. Сравнение последовательностей без выравнивания (I): статистика и мощность. J. Comput. Биол. 16 , 1615–1634 (2009).
MathSciNet
CAS
PubMed
PubMed Central
Google Scholar
Schwende, I.И Фам, Т. Д. Распознавание образов и вероятностные меры в анализе последовательностей без выравнивания. Краткий биоинформ 15 , 354–368 (2014).
Google Scholar
Borozan, I., Watt, S. & Ferretti, V. Интеграция показателей сходства последовательностей на основе и без выравнивания для классификации биологических последовательностей. Биоинф . 31 , 1396–1404 (2015).
CAS
Google Scholar
Дидье, Г., Corel, E., Laprevotte, I., Grossmann, A. & Landès-Devauchelle, C. Локальное декодирование переменной длины и сравнение последовательностей без выравнивания. Теор. Comput. Sci. 462 , 1–11 (2012).
MathSciNet
МАТЕМАТИКА
Google Scholar
Накашима, Х., Нишикава, К. и Оои, Т. Тип сворачивания белка имеет отношение к аминокислотному составу. J. Biochem. 99 , 152–162 (1986).
Google Scholar
Chou, K. C. Некоторые замечания по предсказанию атрибутов белков и псевдоаминокислотному составу (обзор 50-летнего юбилея). J. Theor. Биол. 273 , 236–247 (2011).
CAS
МАТЕМАТИКА
Google Scholar
Мохабаткар, Х., Бейги, М. М., Абдолахи, К. и Мохсензаде, С. Прогнозирование аллергенных белков с помощью концепции псевдоамино-кислотного состава Чоу и подхода машинного обучения. Медицинская химия 9 , 133–137 (2013).
CAS
Google Scholar
Чжун В. и Чжоу С. Молекулярная наука для разработки лекарств и биомедицины. Внутр. J. Molec. Sci . 15 , 20072–20078 (2014).
CAS
Google Scholar
He, P., Wei, J., Yao, Y. & Tie, Z. Новое графическое представление белков и его применение. Physica A 391 , 93–99 (2012).
ADS
CAS
Google Scholar
Randić M. et al. Графическое представление белков. Chem. Ред. 111 , 790–862 (2011).
Google Scholar
Цзян С., Лю В. и Фи К. Х. Графическая теория кинетики ферментов: I. Система реакций в стационарном состоянии, Scientia Sinica 22 , 341–358 (1979).
MathSciNet
МАТЕМАТИКА
Google Scholar
Yao, Y. et al. Анализ сходства / несходства белковых последовательностей. Белки 73 , 864–871 (2008).
CAS
Google Scholar
Куанг, К., Лю, X., Ван, Дж., Яо, Ю. и Дай, К. Статистическая модель последовательностей ДНК с привязкой к положению и ее применение для анализа сходства. MATCH Commun. Математика. Comput. Chem . 73 , 545–558 (2015).
MathSciNet
Google Scholar
Sun, D., Xu, C. & Zhang, Y. Новый метод 2D графического представления белков и его применение. MATCH Commun. Математика. Comput. Chem . 75 , 431–446 (2016).
MathSciNet
Google Scholar
Ся, Х.& Ли, В. Какие свойства аминокислот влияют на эволюцию белка? J. Mol. Evol . 47 , 557–564 (1998).
ADS
CAS
Google Scholar
Qi, Z., Jin, M., Li, S. & Feng, J. Метод картирования белков, основанный на физико-химических свойствах и уменьшении размеров. Comput. Биол. Med. 57 , 1–7 (2015).
Google Scholar
Гутман И.Энергия графика. Ber. Математика. Статист. Sekt. Forschungsz. Грац 103 , 1-22 (1978).
MATH
Google Scholar
Wu, H., Zhang, Y., Chen, W. и Mu, Z. Сравнительный анализ первичных последовательностей белков с графиком энергии. Physica A 43 , 249–262 (2015).
ADS
CAS
МАТЕМАТИКА
Google Scholar
Гутман И., Ли, X. и Чжан, Дж. Энергия графа, в: Анализ сложных сетей. От биологии к лингвистике , (ред. Демер М. и Эммерт-Стрейб Ф.) 145–174 (Wiley-VCH, Weinheim, 2009).
Ли, X., Ши, Й. и Гутман, I. Graph Energy (ред. Ли, X., Ши, Ю. и Гутман) (Springer. New York, 2012).
Замятин А.А. Объем белка в растворе. Прог. Биофиз. Мол. Биол. 24 , 107–123 (1972).
Google Scholar
Чотия, К.Природа доступных и заглубленных поверхностей в белках. J. Mol. Биол. 105 , 1–14 (1975).
Google Scholar
Randić, M. 2D графическое представление белков на основе физико-химических свойств аминокислот. Chem. Phys. Lett. 444 , 176–180 (2007).
ADS
Google Scholar
Паола, Л.Д., Мей, Г., Венере, А. Д. и Джулиани, А. Исследование стабильности димеров через топологию структуры белка. Curr. Белковый пептид Sci . 17 , 30–36 (2016).
Google Scholar
Yu, L., Zhang, Y., Jian, G. & Gutman, I. Классификация данных микрочипов на основе кластеризации K-средних в сочетании с модифицированным отношением одиночного шума к шуму на основе энергии графа, J. Comput. Теор. Nanosci. 14 , 598–606 (2017).
CAS
Google Scholar
Эммерт-Стрейб, Ф., Демер, М. и Ши, Ю. Пятьдесят лет сопоставления графов, выравнивания и сравнения сетей. Информ. Sci . 346–347 , 180–197 (2016).
MathSciNet
МАТЕМАТИКА
Google Scholar
Демер, М., Эммерт-Стрейб, Ф., Чен, З., Ли, X. и Ши, Ю. Математические основы и приложения графовой энтропии , (изд.Демер М. и др.) (Wiley, 2016).
Yu, C., Deng, M. & Yau, S. S. Сравнение последовательностей ДНК новым вероятностным методом. Инф. Sci . 181 , 1484–1492 (2011).
MathSciNet
Google Scholar
Обложка, Т. М. и Томас, Дж. А. Элементы теории информатики , (изд. Wiley, J. & Sons), 2-е издание (Wiley, 1991).
Кульбак, С.& Лейблер, Р. А. Об информации и достаточности. Ann. Математика. Стат . 22 , 79–86 (2015).
MathSciNet
МАТЕМАТИКА
Google Scholar
Yu, C., Cheng, S., He, R. & Yau, S. S. Карта белков: метод сравнения последовательностей без выравнивания, основанный на различных свойствах аминокислот. Ген 486 , 110–118 (2011).
CAS
Google Scholar
Emmert-Streib, F.И Дехмер, М. Обработка информации в сети регуляции транскрипции дрожжей: функциональная устойчивость. BMC Системная биология 3 (2009).
Лам В. и Бахус Ф. Изучение байесовских сетей убеждений: подход, основанный на принципе MDL. Computat. Интелл . 10 , 269–293 (1994).
Google Scholar
Xiao, X. et al. Использование фактора измерения сложности для прогнозирования субклеточного местоположения белка. Аминокислоты 28 , 57–61 (2005).
CAS
Google Scholar
Liao, B., Liao, B., Sun, X. & Zeng, Q. Новый метод анализа сходства и прогнозирования субклеточной локализации белка. Биоинф . 26 , 2678–2683 (2010).
CAS
Google Scholar
Mu, Z., Wu, J. & Zhang, Y. Новый метод анализа сходства / несходства белковых последовательностей. Physica A 392 , 6361–6366 (2013).
ADS
Google Scholar
Чанг, Г. и Ван, Т. Филогенетический анализ белковых последовательностей на основе распределения длины по общей подстроке. Protein J. 30 , 167–172 (2011).
CAS
Google Scholar
Форд, М. Дж. Молекулярная эволюция трансферрина: доказательства положительного отбора у лососевых. Мол. Биол. Evol. 18 , 639–647 (2001).
CAS
Google Scholar
Дэвис П. Л., Баардснес Дж., Койпер М. Дж. И Уокер В. К. Структура и функция антифризовых белков. Phil. Пер. R. Soc. Лондон. B 357 , 927–935 (2002).
CAS
Google Scholar
Думан, Дж., Верлей, Д. и Ли, Н. Сайт-специфичные формы антифриза белка у жука dendroides canadensis. J. Comp. Physiol. B 172 , 547–552 (2002).
CAS
Google Scholar
Graether, S.P. et al. Бета-спиральная структура и свойства связывания льда гиперактивного антифриза насекомого. Nature 406 , 325–328 (2000).
ADS
CAS
Google Scholar
Грейтер, С. П. и Сайкс, Б. Д.Выживание в условиях холода у насекомых, не переносящих заморозки: структура и функция бета-спиральных антифризовых белков. J. Biochem. 271 , 3285–3296 (2004).
CAS
Google Scholar
Altschul, S. F. et al. Gapped LAST и PSI-BLAST: новое поколение программ поиска по базам данных белков. Nucleic Acids Res . 25 , 3389–3402 (1997).
CAS
PubMed
PubMed Central
Google Scholar
Яу, С., Yu, C. & He, R. Карта белков и ее применение. клетки ДНК. Биол. 27 , 241–250 (2008).
CAS
Google Scholar
Xu, C., Sun, D., Liu, S. & Zhang, Y. Анализ последовательности белка путем включения модифицированной игры хаоса и физико-химических свойств в общий псевдоаминокислотный состав Чжоу. J. Theor. Биол. 406 , 105–115 (2016).
CAS
Google Scholar
|
|
Мета-функциональные сигнатуры белков на основе сочетания последовательности, структуры, эволюции и информации о свойствах аминокислот
Abstract
Функция белка опосредуется различными аминокислотными остатками, как их положениями, так и типами, в последовательности белка. Некоторые аминокислоты отвечают за стабильность или общую форму белка, играя косвенную роль в функции белка.Другие играют функционально важную роль как часть активных или связывающих сайтов белка. Для данной белковой последовательности остатки и степень их функциональной важности можно рассматривать как сигнатуру, представляющую функцию белка. Мы разработали комбинацию подходов к прогнозированию функций, основанных на знаниях и биофизике, для выяснения отношений между структурными и функциональными ролями отдельных остатков и положений. Такая метафункциональная сигнатура (MFS), которая представляет собой набор непрерывных значений, представляющих функциональную значимость каждого остатка в белке, может использоваться для более детального изучения белков с известной функцией и для помощи в экспериментальной характеристике белков неизвестного происхождения. функция.Мы демонстрируем превосходную производительность MFS в прогнозировании функциональных сайтов белков, а также представляем четыре реальных примера применения MFS в широком диапазоне настроек для выяснения взаимосвязей между последовательностью белка, структурой и функцией. Наши результаты показывают, что подход MFS, который может объединить несколько источников информации, а также дать биологическую интерпретацию каждого компонента, значительно облегчает понимание и характеристику функции белка.
Сведения об авторе
Белки — это основные строительные блоки и функциональные молекулы клетки.Функция опосредуется конкретными аминокислотными остатками в последовательности белка в зависимости как от их положения, так и от типа. Белки традиционно описываются как последовательность аминокислот и, если они известны, экспериментально определенные координаты этой ковалентно связанной цепи. Здесь мы предлагаем расширить описание белка, включив в него количественную меру функциональной важности каждой составляющей аминокислоты. Результирующая сигнатура для белковой последовательности или структуры называется ее метафункциональной сигнатурой (MFS).Мы представляем ансамбль методов, основанных на знаниях и биофизике, которые используют различные типы доказательств для функциональной важности, в качестве автоматизированного общедоступного инструмента для построения такой MFS. Мы используем два набора контрольных данных, чтобы показать, что MFS можно использовать для идентификации функционально важных остатков только на основе структуры или последовательности белка. Наконец, мы оцениваем четыре разнообразных реальных биологических вопроса, чтобы продемонстрировать способность MFS дать представление о структурных и функциональных ролях отдельных остатков и положений, используя взаимосвязи между последовательностью белка, структурой и функцией.
Образец цитирования: Wang K, Horst JA, Cheng G, Nickle DC, Samudrala R (2008) Мета-функциональные сигнатуры белков на основе комбинации информации о последовательности, структуре, эволюции и аминокислотных свойствах. PLoS Comput Biol 4 (9):
e1000181.
https://doi.org/10.1371/journal.pcbi.1000181
Редактор: Расс Б. Альтман, Стэнфордский университет, Соединенные Штаты Америки
Поступила: 15 апреля 2008 г .; Принята к печати: 7 августа 2008 г .; Опубликовано: 26 сентября 2008 г.
Авторские права: © 2008 Wang et al.Это статья в открытом доступе, распространяемая в соответствии с условиями лицензии Creative Commons Attribution License, которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Финансирование: Эта работа была поддержана стипендией Searle Scholar, наградой Национального научного фонда (NSF) CAREER, грантом NSF DBI-0217241, а также грантами Национальных институтов здравоохранения GM068152-01 и F30DE017522-02.
Конкурирующие интересы: Авторы заявили об отсутствии конкурирующих интересов.
Введение
Огромные объемы последовательностей и структурных данных генерируются высокопроизводительными технологиями. Функциональные аннотации не охарактеризованных последовательностей и структур значительно отстают. Время и стоимость экспериментальных методов, необходимых для исследования функции всех не охарактеризованных белков, непомерно высоки. Поэтому вычислительные средства становятся все более полезными и популярными для прогнозирования и аннотирования функций для огромного количества данных последовательности и структуры [1], [2].
Однако предсказание функции белка само по себе является сложной задачей для формулирования, так как трудно определить функцию [2], [3]. Различные схемы функционального определения (такие как Комиссия по ферментам [4], Онтология генов [5] и суперсемейство SCOP [6]) были разработаны на протяжении многих лет и касались различных аспектов функции белков. Вместо того, чтобы принять существующую схему функционального определения, мы предложили исследовать роль отдельных аминокислотных остатков в функции белка, независимо от используемых схем функционального определения.В таких случаях функция белка может быть представлена просто как ряд количественных значений, каждое из которых указывает на функциональную важность соответствующего аминокислотного остатка в последовательности или структуре белка. Чтобы вычислить количественные значения для каждого остатка, мы использовали комбинированный подход, метафункциональную сигнатуру (MFS), которая учитывает индивидуальные оценки из различных алгоритмов прогнозирования функций и генерирует совокупную оценку для каждого аминокислотного остатка в данном белке. .В настоящее время наш протокол генерации подписи состоит из следующих четырех типов оценок для четырех различных типов информации: (1) сохранение последовательности, (2) эволюционное сохранение, (3) структурная стабильность и (4) тип аминокислоты. Все эти оценки генерируются с помощью концептуально простых и легко реализуемых алгоритмов (описанных ниже), а их совместное использование превосходит сложные алгоритмы, использующие только один источник информации.
Консервация последовательности — один из наиболее часто используемых методов измерения функциональной важности отдельных аминокислот.Аминокислотные остатки с более консервативными моделями вариаций обычно более важны для сохранения функции белка. Эта концепция часто используется для идентификации функциональных областей белков путем построения нескольких выравниваний между последовательностью-мишенью и всеми ее гомологами последовательностей, а затем анализа степени сохранения последовательности среди каждого сайта выравнивания. На протяжении многих лет предлагались различные меры по сохранению последовательностей, различающиеся по сложности и изощренности [7].Простейшими мерами сохранения последовательности являются оценка энтропии и ее варианты [8] — [13]. Более сложные измерения [14] — [16] включают другую информацию, такую как попарное сходство аминокислот, физико-химические свойства и теоретические профили последовательностей, в схемы оценки. Пакет программ AL2CO включает девять различных схем оценки, но эти оценки имеют тенденцию коррелировать друг с другом [17]. Недавно было также показано, что мера дивергенции Дженсена-Шеннона улучшает предсказание функционально важных остатков, и что учет сохранения в последовательно соседних сайтах еще больше повышает точность [18].Ранее мы продемонстрировали, что мера относительной энтропии, которая включает фоновые частоты аминокислот, может лучше предсказать функциональные сайты, чем простые меры энтропии [19]. Кроме того, мы обнаружили, что включение частот аминокислот, оцененных с помощью скрытых марковских моделей (HMM), дополнительно улучшает характеристики меры относительной энтропии [19]. В текущем исследовании мы используем меру сохранения последовательности, полученную из HMM (HMM_rel_ent), как один из компонентов нашего протокола генерации метафункциональной подписи.
В дополнение к сохранению последовательности, мы также включаем информацию о сохранении эволюции в метафункциональную сигнатуру. Многие исследования показали, что использование филогенетических отношений между группой эволюционно связанных последовательностей помогает точно предсказать функциональные сайты. Метод Evolutionary Trace, один из первых и наиболее успешных из таких методов, анализирует паттерны вариации остатков внутри и между подсемействами белков в результате множественных выравниваний, отображает важные остатки в структуру белка и количественно оценивает важность остатков [20], [21].Дальнейшее развитие метода эволюционного следа позволяет количественно ранжировать важность остатков, комбинируя использование эволюционной информации и энтропийных мер [22], [23]. Точно так же метод ConSurf конструирует филогенетические отношения из группы схожих последовательностей, вычисляет оценку сохранения с помощью байесовского метода или метода максимального правдоподобия и отображает информацию о сохранении на поверхности белка [24], [25]. Кроме того, исследование Soyer et al. использовали сайт-специфические эволюционные модели, которые предполагали различную матрицу замен для каждого сайта, для обнаружения функциональных сайтов белка [26].La et al. использовали эволюционные отношения между фрагментами последовательностей (филогенетические мотивы) для определения функциональных сайтов белков [27]. дель Соль Меса и др. представили несколько автоматизированных методов, которые делят данное семейство белков на подсемейства и ищут остатки, определяющие специфичность [28]. Общность всех этих методов заключается в том, что взаимосвязи последовательностей анализируются на основе топологии эволюционного дерева, что обеспечивает дополнительный уровень информации вместо того, чтобы полагаться только на несколько выравниваний последовательностей.Здесь мы предлагаем новый метод, называемый оценкой отношения состояния к шагу (SSR), для измерения эволюционного сохранения. На основе заданных множественных выравниваний мы строим дерево максимальной экономии и анализируем модели вариаций от корня дерева (теоретическая наследственная последовательность) до листа дерева (последовательности в нескольких выравниваниях), чтобы создать оценку для каждого аминокислотного остатка. . Оценка SSR — простой, но эффективный способ измерения эволюционной сохранности.
Оценки функциональной сигнатуры также могут быть получены с помощью методов, основанных на биофизике, с использованием экспериментально определенных или предсказанных с помощью вычислений структур белков.Например, недавнее исследование продемонстрировало, что дестабилизирующие области в белковых структурах часто могут быть использованы для получения ценной информации для функциональных выводов и идентификации функциональных сайтов [29]. Для данной структуры и данного положения мы предлагаем мутировать остаток дикого типа на 19 других аминокислот и рассчитать их оценки структурной стабильности, которые, в свою очередь, можно использовать для присвоения оценки каждому остатку в белке. Следовательно, эти оценки также могут служить компонентом прогнозирования функции белков.Ранее мы разработали специфичную для остатков функцию различения вероятностей для всех атомов (RAPDF) [30], которая собирает статистику из базы данных экспериментальных структур для оценки и выбора «ложных» структур, которые с большей вероятностью будут похожи на экспериментально полученные структуры. RAPDF был оптимизирован и усовершенствован в последние годы для предсказания структуры белков [31] — [33]. Здесь мы дополнительно расширили RAPDF для оценки мутаций остатков на основе остатков. Каждый остаток в данном белке был изменен на одну из 19 альтернативных аминокислот, создав новые структуры, которые были дополнительно оптимизированы по топологии (через перестройку боковой цепи) и максимизированы для стабильности (через глобальное возмущение конформации).В нашем текущем протоколе генерации MFS мы использовали две функции оценки на основе RAPDF (RAPDF_spread и RAPDF_dif), чтобы измерить, как все мутировавшие структуры отклоняются друг от друга и как экспериментально определенная структура отличается от мутированных структур, что представляет потенциальное влияние на стабильность для положение и для встречающегося в природе остатка, соответственно. Эти оценки отделяют остатки, сохраняемые для структуры от функции.
Дополнительным компонентом метафункциональной сигнатуры является информация о типе аминокислот, таких как гистидин и цистеин, которые с большей вероятностью располагаются в функциональных сайтах, чем другие аминокислоты.Однако такая «априорная вероятность» для функционального сайта явно не моделируется и не включается в большинство современных алгоритмов прогнозирования функционального сайта. В нашем протоколе создания MFS мы использовали 19 бинарных переменных (все, кроме аланина), чтобы представить идентичность аминокислот для каждой позиции в данном белке. Мы также исследовали, может ли явное использование информации об аминокислотах (например, AAType), в отличие от неявного использования (например, посредством вычисления относительной энтропии), предоставить дополнительную информацию и повысить производительность.
Учитывая сложность определения и идентификации функциональных сайтов белков, очевидно, что ни один метод не всегда будет работать для сбора всей информации о функциональных сайтах белка. Поэтому несколько групп начали использовать информацию из различных источников, особенно информацию, полученную из структур, чтобы давать более точные прогнозы. Работа Chelliah et al. показал, что различение структурных и функциональных ограничений для аминокислотных остатков приводит к лучшему предсказанию сайтов взаимодействия с белками [34].Мы показали, что, рассматривая как структурные, так и функциональные ограничения эволюции белков, мы можем лучше идентифицировать функциональные сайты и сигнатуры [35], [36]. Недавно Петрова и соавт. показали, что интеграция семи выбранных последовательностей и структурных особенностей в каркас машины опорных векторов (SVM) может улучшить идентификацию каталитических сайтов [37]. Кроме того, Fischer et al. интегрированная консервация последовательности, распределение аминокислот, предсказанная вторичная структура и относительная доступность растворителя в структуре плотности вероятности и показали, что при 20% чувствительности интегрированный метод приводит к 10% увеличению точности по сравнению с неинтегрированными методами предсказания каталитических остатков из Атлас каталитического участка и записи САЙТА PDB [38].Youn et al. исследовали различные особенности для различения каталитических остатков от некаталитических в новых структурных складках и показали, что мера сохранения последовательности, мера структурной консервации, степень уникальности структурной среды остатка, доступность растворителя и гидрофобность остатка являются лучшими предикторами. каталитических центров [39]. Другие аналогичные исследования также включали от десятков до сотен функций в структуру машинного обучения для идентификации каталитических сайтов [40], [41].В целом, предыдущая работа предполагает большую ценность использования нескольких комплементарных последовательностей и структурных компонентов для оценки каталитических центров. В отличие от этих подходов, которые в значительной степени основывались на алгоритмах машинного обучения, в текущем исследовании мы стремимся объединить несколько источников информации о последовательности, структуре, эволюции и типе аминокислот вместе с помощью простой модели логистической регрессии для прогнозирования функций, включая как каталитические сайты, так и сайты связывания. Основным преимуществом регрессионной модели является то, что каждый компонент может быть связан с биологически значимой интерпретацией, и что отдельные баллы для белка можно изучать вручную, чтобы получить дополнительную информацию о различных аспектах функции белка, которые недоступны, когда много компонентов брошены в сложную структуру машинного обучения.Мы сравниваем подход MFS с несколькими другими алгоритмами прогнозирования функционального сайта, предлагаем улучшения нашего подхода, приводим примеры широкого определения функции, оцениваемой MFS, и обсуждаем, как различные компоненты MFS могут использоваться для понимания биологической функции на четырех реальных примерах.
Методы
Компоненты метафункциональных сигнатур
Оценка сохранения последовательности.
Мы провели поиск каждой последовательности запросов в базе данных Uniref90 [42], используя три итерации программы PSI-BLAST [43], и построили несколько сопоставлений.Затем мы составили модель HMM, используя пакет HMMER [44], и рассчитали относительную энтропию положения, используя частоты аминокислот, оцененные с помощью модели HMM.
Оценка HMM_rel_ent была рассчитана следующим образом: p i ( i = 1,…, 20) представляет частоту эмиссии аминокислот, оцененную с помощью модели HMM, а p ib представляет заданную частоту аминокислотного фона. в karlin.c программного пакета BLAST [43].
Эволюционная оценка сохранения.
Используя множественные выравнивания, сгенерированные на предыдущем шаге, мы построили филогенетические деревья с методами максимальной экономии, используя программу protpars в программном пакете PHYLIP [45]. Когда существовало несколько столь же экономных деревьев, мы использовали первое дерево. Затем для каждой выровненной позиции мы вычисляем отношение состояния к шагу (SSR), где N, , состояние , — это количество типов остатков в данной позиции выравнивания, а N, шаг , , — общее количество изменений типа остатка в положение, выведенное из корня дерева.
Оценка структурной устойчивости.
В качестве индикатора структурной стабильности мы использовали показатель вероятностно-дискриминирующей функции для всех атомов (RAPDF). Оценка RAPDF основана на условной вероятности того, что конформация является нативной, с учетом набора межатомных расстояний. Подробная формулировка шкалы RAPDF описана в [30], [31]. Первоначальная версия этой функции использовалась в качестве ключевого компонента наших методов предсказания структуры белка, которые хорошо работают в экспериментах по слепому предсказанию CASP [33], [46].В текущем исследовании мы использовали модифицированную версию оценки RAPDF [32], RAPDF с 37 ячейками, с использованием интервалов расстояний с интервалом 0,5 Å (вместо интервала 1 Å в исходной формуле).
Для каждого аминокислотного остатка в данной структуре белка мы сначала мутировали аминокислоту до одной из 19 альтернативных аминокислот и использовали программу генерации боковой цепи SCWRL [47], чтобы перестроить боковую цепь мутированной аминокислоты. Мы применили протокол минимизации энергии ENCAD [48] в качестве промежуточного шага (необязательного в программном обеспечении MFS), чтобы минимизировать стерические помехи.Затем мы вычислили значения RAPDF с помощью модифицированной версии потенциальной программы в программном пакете RAMP, которая использует 37 интервалов расстояния для статистического вывода [32]. Затем из набора из 20 значений RAPDF для аминокислоты дикого типа и 19 альтернативных аминокислот мы составили две разные итоговые оценки.
Первая суммарная оценка — это оценка RAPDF_spread, которая представляет собой стандартное отклонение оценок RAPDF для 20 мутированных структур, которые отличаются одним остатком, и рассчитывается как
Вторая суммарная оценка — это оценка RAPDF_dif, которая рассчитывается как где S RAPDF, wt — это значение RAPDF для структуры дикого типа.Оценка RAPDF_dif вычисляет разницу между структурой дикого типа и средним значением всех 20 возможных структур, в то время как оценка RAPDF_spread оценивает все 20 оценок как распределение и не связано с идентичностью аминокислоты дикого типа. Обе оценки измеряют различные аспекты структурной стабильности, вызванной мутациями аминокислот: оценка RAPDF_dif оценивает влияние аминокислоты дикого типа на стабильность, тогда как оценка RAPDF_spread оценивает потенциальное влияние этой позиции.
Оценка по типу аминокислот.
Поскольку разные аминокислоты могут иметь различное распределение в функционально важных и неважных сайтах (априорная вероятность того, что аминокислота является функционально важной), мы также ввели в нашу модель набор фиктивных переменных, представляющих аминокислотную идентичность остатка, являющегося считается. 19 оценок, S атип, 2 ,…, S атип, 20 , все являются бинарными переменными (принимающими значение 1 или 0) и указывают, присутствует ли соответствующая аминокислота или нет (AAType).
Обработка несоответствия между структурой последовательности и положением.
Мы использовали наборы данных функциональных сайтов на основе структуры для оценки производительности наших методов. Многие файлы PDB содержат разрывы цепочки, поэтому использование записей ATOM в схемах оценки на основе последовательностей неразумно, поскольку сгенерированные множественные выравнивания могут быть неточными, особенно при наличии больших разрывов цепочки. В нашем методе MFS две оценки подписи на основе последовательности (HMM_rel_ent, SSR) генерируются с использованием записей SEQRES файлов PDB; следовательно, необходим перевод этих основанных на SEQRES координат в основанные на ATOM координаты.Для этого мы выполнили глобальное попарное выравнивание последовательности на основе ATOM и последовательности на основе SEQRES с использованием алгоритма Нидлмана-Вунша, реализованного в программном пакете EMBOSS [49]. Затем мы проанализировали каждую выровненную позицию, чтобы решить проблему несоответствия SEQRES-ATOM: пробелы в выравнивании указывают на разрывы цепи в записях ATOM, в то время как несогласованные остатки в выравнивании представляют собой мутировавшие остатки при кристаллизации структуры. Мы отмечаем, что хотя глобальное выравнивание последовательностей обычно работает хорошо, могут быть случаи, когда очень большие разрывы цепи препятствуют точному выравниванию; в этих случаях внешние инструменты, такие как сервер S2C (http: // dunbrack.fccc.edu/Guoli/s2c/index.php) может использоваться вместе с файлами PDB для связи последовательности с координатами с данными, полученными из файлов в формате XML. Оценки сигнатур, сгенерированные из последовательности на основе SEQRES, затем могут быть присвоены соответствующим аминокислотным остаткам на основе ATOM в файле PDB.
Построение регрессионных моделей.
После того, как мы сгенерировали оценки HMM_rel_ent, SSR, RAPDF_spread, RAPDF_dif и AAType, мы затем сопоставили данные с известными функциональными сайтами, используя следующую модель логистической регрессии: где p — это вероятность того, что позиция является функционально важной позицией, , –, f — это параметры модели, а ε — погрешность.Подбор модели, проверка модели, оценка производительности и эксперименты по перекрестной проверке проводились в программной среде программирования STATA версии 9.2 (Колледж-Стейшн, Техас).
Оценка эффективности идентификации функционального узла
Мы использовали набор данных Thornton [50] и набор данных Lovell [34], чтобы оценить эффективность MFS и его вариантов при идентификации функциональных сайтов из белковых структур. Набор данных Thornton содержит 1546 активных сайтов ферментов из 508 белков, а набор данных Lovell содержит 1137 функциональных сайтов из 243 белков.Мы оценили эффективность функциональной идентификации сайта по двум критериям, которые использовались в предыдущих исследованиях [19]. Первым критерием является оценка ROC, которая оценивает, как количественные прогнозы функциональной важности соотносятся с бинарными определениями того, является ли сайт функциональным. Эта оценка рассчитывается как площадь под кривой путем построения графика соотношения ложноположительных результатов и истинных положительных результатов в диапазоне пороговых значений. Второй критерий — это 10 самых успешных результатов, которые подсчитывают, сколько из 10 лучших оценочных остатков в данном белке также являются остатками активного сайта.Для данного набора данных сумма 10 лучших результатов для всех белков используется для оценки производительности различных алгоритмов. Кроме того, мы также рассчитали специфичность и количество ложноположительных результатов для каждого белка при достижении 20% чувствительности. Предполагая, что TP, TN, FP и FN представляют собой истинно положительные, истинно отрицательные, ложноположительные и ложно отрицательные прогнозы, соответственно, чувствительность относится к TP / (TP + FN), точность относится к TP / (FP + TP) и частота ложных срабатываний относится к FP / (FP + TN).Для методов MFS и SeqonlyMFS мы применили эксперименты с пятикратной перекрестной проверкой, чтобы оценить их эффективность: весь набор данных был разделен на пять частей, и во время каждой перекрестной проверки 80% белков использовались для обучения модели, что было затем протестировали оставшиеся 20% белков.
Мы оценили эффективность метода MFS по сравнению с двумя широко используемыми программами идентификации функциональных сайтов для белковых структур: сервером Evolutionary Trace (http: // mammoth.bcm.tmc.edu/report_maker) и сервер ConSurf (http://consurf.tau.ac.il). Мы использовали идентификатор PDB для запроса наборов данных Thornton и Lovell, используя оба сервера со всеми параметрами по умолчанию, и собрали выходные файлы ZIP с сервера ET и выходные файлы «оценки сохранения аминокислот» с сервера ConSurf. Некоторые белки генерируют сообщения об ошибках или не могут обрабатываться ни одним из серверов, поэтому они не были включены в наш анализ. Затем мы использовали значение «rho ET score» из файла оценки ET и значение сохранения из файла оценки ConSurf, чтобы оценить производительность этих методов с помощью ROC и 10 лучших результатов.Сервер ET генерирует много равных оценок (обычно намного больше 10) для остатков с наивысшей оценкой; Таким образом, рейтинг 10 лучших совпадений не использовался для ET в нашем сравнительном анализе.
Для каждого метода мы также создали файлы модифицированной структуры PDB, в которых поле температуры было заменено прогнозируемыми оценками функциональной важности. Затем эти структуры были визуализированы с использованием программного обеспечения UCSF chimera [51], так что цвет каждого остатка представляет собой значение оценки функциональной важности.Визуальный осмотр сгенерированных структур помогает понять, как и почему каждый метод работал или не удался.
Реализация веб-сервера для создания MFS
Мы реализовали протокол генерации MFS как веб-сервер, доступный по адресу http://protinfo.compbio.washington.edu/mfs. Входные данные для этого сервера представляют собой последовательность из одной цепочки или структуру в формате FASTA или PDB соответственно, а выходными данными является прогнозируемая оценка MFS для каждого остатка в структуре.Кроме того, когда предоставляется входная структура, создается новый файл структуры с полем температурного фактора, замененным оценками MFS, чтобы обеспечить визуальный контроль функционально важных областей с помощью программного обеспечения для молекулярной графики. Если файл структуры содержит много разрывов цепи в записях ATOM, пользователь может дополнительно отправить полную последовательность, чтобы можно было сгенерировать более точное выравнивание последовательностей для белка запроса. Если пользователи отправляют только информацию об аминокислотной последовательности, то для прогнозирования функциональных сайтов будет использоваться протокол генерации SeqonlyMFS.Для белка среднего размера с 200 остатками вычисление для SeqonlyMFS может быть выполнено в течение одного часа, в то время как вычисление для MFS на основе структуры может быть выполнено в течение одного дня, когда очередь на обработку не занята. Этот сервер будет постоянно обновляться, когда наш протокол генерации MFS будет дорабатываться и улучшаться. Автономный исходный код, используемый для создания MFS, также можно загрузить по тому же URL-адресу.
Результаты
Вклад компонентов метафункциональной сигнатуры в идентификацию функционального сайта
Оценка производительности наших протоколов метафункциональной сигнатуры (MFS) потребовала от нас использования «золотого стандарта» набора данных функциональных сайтов белков с известной структурой.Мы не использовали записи «SITE» в файлах PDB или записи «ACT_SITE» в файлах Swiss-Prot, потому что эти аннотации, как правило, нечетко определены и содержат высокий уровень ошибок и низкий уровень охвата [50]. Вместо этого мы использовали набор данных Thornton [50] и набор данных Lovell [34], которые использовались в предыдущих экспериментах [19], [36]. Набор данных Thornton содержит аннотированные вручную активные центры ферментов, извлеченные из первичной литературы; набор данных Lovell содержит вручную скомпилированные сайты связывания лигандов на основе литературы.Для оценки производительности мы использовали показатель ROC и рейтинг топ-10, как описано ранее [19]. Чтобы исследовать добавленную стоимость каждого компонента метафункциональных сигнатур, мы сравнили характеристики дополнительных компонентов MFS: сохранение последовательности (HMM_rel_ent), эволюционное сохранение (SSR), тип аминокислоты (AAType), структурная стабильность положения (RAPDF_spread) ) и остаточной структурной стабильности (RAPDF_dif) (рисунок 1). Последовательное включение каждого компонента улучшает производительность.MFS, использующая максимальное количество компонентов, имеет лучшую производительность при прогнозировании функциональных сайтов.
Рис. 1. Точность функциональной идентификации сайта в наборах данных Thornton и Lovell несколькими методами, которые используют только информацию о последовательности (HMM_rel_ent), затем с добавлением эволюционной информации (HMM_rel_ent + SSR) с последующим добавлением информации о типе аминокислоты (HMM_rel_ent + SSR + AAType) и, наконец, с дополнительной структурной информацией (MFS).
Для оценки производительности использовались баллы ROC и 10 лучших результатов. Четыре метода имеют возрастающую точность, демонстрируя важность объединения информации о последовательности, структуре, эволюции и типе аминокислот при функциональной характеристике белков.
https://doi.org/10.1371/journal.pcbi.1000181.g001
Высокая корреляция между компонентами (независимыми переменными) в линейной модели будет иметь тенденцию дестабилизировать параметры модели и давать ошибочную статистическую значимость.Чтобы выяснить, есть ли у наших моделей MFS такие проблемы, мы проверили коэффициент инфляции дисперсии (VIF). VIF — это мера для каждой независимой переменной, позволяющая оценить, как коллинеарность переменных влияет на точность оценки параметров. Оценка VIF выше 10 обычно указывает на проблемные модели. Мы обнаружили, что все оценки VIF для параметров в моделях MFS при применении к обоим наборам данных меньше 4, что указывает на то, что наши модели не страдают от проблем коллинеарности. Кроме того, мы рассчитали парные коэффициенты корреляции между оценкой HMM_rel_ent, оценкой SSR, оценкой RAPDF_spread и оценкой RAPDF_dif для обоих наборов данных (таблица 1).Мы обнаружили, что максимальное абсолютное значение коэффициента корреляции составляет 0,45 между оценками HMM_rel_ent и SSR. Следовательно, каждый компонент протокола MFS предоставляет дополнительную и преимущественно ортогональную информацию, и их можно использовать индивидуально для оценки различных аспектов функции.
Таблица 1. Коэффициенты корреляции нескольких компонентов метода MFS в наборе данных Thornton (ячейки в верхнем правом треугольнике таблицы) и в наборе данных Ловелла (нижний левый треугольник), соответственно.
https://doi.org/10.1371/journal.pcbi.1000181.t001
Сравнительный анализ характеристик метафункциональной сигнатуры
Было создано несколько веб-серверов, которые присваивают количественные оценки функционально важным аминокислотным остаткам и сопоставляют эти оценки со структурами белков для идентификации пространственных кластеров важных остатков. Мы сравнили производительность MFS с двумя такими веб-серверами, сервером Evolutionary Trace (ET) и сервером ConSurf.Сервер ET реализует метод, который объединяет эволюционную и энтропийную информацию для ранжирования каждого остатка по его функциональной важности [23], в то время как метод ConSurf использует филогенетическую информацию для измерения сохранения остатка [24]. Хотя и методы ET, и методы ConSurf сопоставляют оценки со структурами белков, эти методы не используют явным образом структурную информацию при расчете функциональной важности. Поэтому для сравнения мы также использовали метод SeqonlyMFS, который не использует структурную информацию.
Для сравнения этих методов мы использовали те же наборы данных и показатели производительности, что и в предыдущем разделе. Однако, поскольку сервер ET и сервер ConSurf выдавали сообщения об ошибках или не могли обрабатывать некоторые белки, мы сосредоточили наш анализ на белках 453/508 в наборе данных Thornton и на белках 226/243 в наборе данных Lovell, для которых оба сервера генерировали выходные данные (рис. 2). Кроме того, мы не подсчитывали топ-10 результатов для сервера ET, потому что для любого данного белка этот сервер обычно генерирует намного больше, чем 10 равных оценок, связанных на первом месте.Мы обнаружили, что MFS и SeqonlyMFS превосходят оба сервера при сравнении их показателей ROC: для сравнения SeqonlyMFS и ET P-значения знакового теста составили 1,2e-25 и 4,4e-15 для наборов данных Thornton и Lovell соответственно; для сравнения SeqonlyMFS и ConSurf значения P составили 1,4e-39 и 1,3e-16 соответственно. Кроме того, SeqonlyMFS и MFS сгенерировали значительно больше попаданий в топ-10, чем сервер ConSurf для обоих наборов данных. Отметим, что в реальных приложениях более важно оценивать производительность, когда даются только самые надежные прогнозы; поэтому мы также сравнили показатель точности и частоту ложных срабатываний при достижении 20% чувствительности для каждого белка.По обоим параметрам MFS по-прежнему имеет лучшую производительность среди всех методов (рисунок 2). Наконец, поскольку каждый белок может иметь различное количество функциональных сайтов, сумма первых 10 совпадений для всех белков может не быть оптимальной мерой ожидаемой производительности для данного белка. Поэтому мы рассчитали чувствительность каждого метода для каждого белка. Для набора данных Thornton средние значения чувствительности для всех белков составляют 67,0%, 62,5% и 33,7% для MFS, SeqonlyMFS и ConSurf соответственно.Для набора данных Lovell средние значения чувствительности составляют 70,0%, 66,9% и 40,8% соответственно. В целом, по сравнению с методами, использующими только один источник информации, подход MFS, объединяющий несколько источников информации, может дать улучшенную производительность при прогнозировании функционально важных остатков.
Рисунок 2. Сравнение производительности метода MFS, метода SeqonlyMFS (HMM_rel_ent + SSR + AAType), метода эволюционной трассировки и метода ConSurf с наборами данных Thornton и Lovell.
В сравнении используются только те белки, для которых методы Evolutionary Trace и ConSurf могут давать прогнозы. Для сравнения производительности используются четыре показателя, в том числе: оценки ROC, точность, когда порог чувствительности установлен на 20%, частота ложных срабатываний, когда порог чувствительности установлен на 20%, и 10 лучших результатов. ET используется только при вычислении оценки ROC, но не в другом сравнительном анализе, поскольку он дает много равных оценок для остатков с наивысшей оценкой. И методы MFS, и SeqonlyMFS имеют лучшую производительность, чем методы, использующие только один тип информации.
https://doi.org/10.1371/journal.pcbi.1000181.g002
Приложения метафункциональных подписей
Метод MFS можно рассматривать как инструмент для определения функции белка как серии количественных значений. В качестве альтернативы, при рассмотрении каждого компонента MFS также можно рассматривать как несколько векторов с одинаковыми размерами. В предыдущих разделах мы продемонстрировали применение MFS для функциональной идентификации сайтов. Здесь мы также демонстрируем использование MFS в других типах задач вычислительной биологии на четырех примерах.
Идентификация биологических механистических остатков путем сопоставления оценок MFS с белковыми структурами.
Было доказано, что сопоставление определенной группы остатков в последовательности белка со структурой белка является мощным способом изучения функции белка, поскольку визуальный осмотр человека часто может выявить закономерности кластеризации остатков и помочь в интерпретации взаимосвязей структура-функция . Мы применили этот подход, чтобы изучить, как и почему работает метод MFS, сравнив образцы высокоуровневого картирования остатков, сгенерированные различными методами.
Орнитиндекарбоксилаза .
Мы использовали прогнозируемые баллы функциональной важности для орнитиндекарбоксилазы (идентификатор PDB 1ord-A) в качестве примера, чтобы проиллюстрировать различную производительность четырех методов: MFS, SeqonlyMFS, ET и ConSurf. Структуры представлены в виде лент, с тремя функциональными каталитическими сайтами (223H-316D-355K), отмеченными сферами, а все остатки окрашены в соответствии с их прогнозируемой оценкой функциональной важности (рис. 3A). Для этого белка 3, 2 и 0 функциональных сайтов правильно идентифицированы в первой десятке совпадений методами MFS, SeqonlyMFS и ConSurf соответственно (ET идентифицирует 3 сайта в своих топ-58 совпадениях из-за многих связанных оценок).Поэтому подробный анализ этих структур поможет нам понять, чем и почему методы различаются по своей производительности.
Рис. 3. Различная прогностическая эффективность метода MFS, метода SeqonlyMFS, сервера эволюционной трассировки и сервера ConSurf на двух примерах.
Структура орнитиндекарбоксилазы (A) (идентификатор PDB 1ord-A) и целлобиогидролазы (B) (идентификатор PDB 1cel-A) показана на ленточных изображениях с функциональными сайтами (223H-316D-355K в 1ord-A). A, 212E-214D-217E-228H в 1cel-A) в виде сфер.Каждый остаток окрашен в соответствии с прогнозируемой оценкой функциональной важности, причем цвет меняется с красного на белый и на синий по мере уменьшения оценки. Для 1ord-A (A) и MFS, и SeqonlyMFS хорошо работают при присвоении наивысших оценок функциональным сайтам. Тем не менее, ET и ConSurf также присваивают высокие баллы ближайшим остаткам в окружающей полости, поэтому функциональные сайты не появляются в списках первой десятки совпадений, созданных этими методами. Для 1cel-A (B) все методы на основе последовательностей могут назначать относительно высокие баллы функциональным сайтам (разные оттенки красного цвета), но только метод MFS, который использует структурную информацию, может повысить оценки функциональных сайтов. выше (более интенсивный красный цвет), чтобы появиться в списке 10 лучших результатов.
https://doi.org/10.1371/journal.pcbi.1000181.g003
Ортининдекарбоксилаза имеет три структурных домена: N-концевой «крыловидный» домен (внизу слева на рисунке), RLP-зависимая трансфераза. домен, который содержит большую полость с каталитической триадой внутри и небольшой C-концевой α + β домен, который частично закрывает полость (верхний структурный домен на рисунке). Как методы ET, так и ConSurf присваивают высокие баллы (показаны красным и светло-красным цветом) многим остаткам вокруг полости белка.Однако три активных центра не получают наивысших оценок этими двумя методами, поэтому методы ET и ConSurf не могут отличить эти остатки от других остатков в той же полости. В таких случаях, хотя кластер высоко оцениваемых остатков можно различить визуально, химически функциональные сайты по-прежнему не могут быть легко выведены этими двумя методами. Однако, поскольку как методы MFS, так и методы SeqonlyMFS используют информацию, основанную на типе аминокислот, они могут генерировать более высокие баллы для функциональных сайтов, наблюдаемых в наборах тестов (в нашей модели гистидин, аспарагиновая кислота и лизин имеют более высокие оценки. вкладов, чем другие типы остатков), что приводит к лучшей идентификации биологически механистических функциональных сайтов.
Целлобиогидролаза .
Вторым примером является целлобиогидролаза (идентификатор PDB: 1cel-A), которая имеет сэндвич-образную складку, содержащую несколько нитей на двух листах (рис. 3B). Четыре функциональных сайта (212E-214D-217E-228H) последовательно и пространственно близки друг к другу. Только метод MFS может правильно идентифицировать 3 из 4 функциональных сайтов для этого белка в списке топ-10, в то время как методы SeqonlyMFS и ConSurf не могут идентифицировать ни одного (ET идентифицирует 3 сайта в своих 52 лучших сайтах из-за многих связанных баллов).Чтобы упростить визуальный осмотр, мы раскрасили структуру так, чтобы только остатки с относительно высокой оценкой имели разные оттенки красного, а все остальные остатки были синими. (Например, для метода SeqonlyMFS четыре функциональных сайта показаны белым, голубым, светло-красным и красным цветом соответственно, что указывает на то, что они имеют все более высокие оценки функциональной важности.) Ни один из методов, основанных на последовательностях, не может определить истинное функциональные сайты, потому что последовательности, которые соответствуют этой конкретной структурной складке, являются высококонсервативными, и многие остатки в двух листах имеют относительно высокие оценки консервативности.Однако, поскольку все остатки в двух бета-листах находятся в непосредственной близости друг от друга, оценки RAPDF с большей вероятностью будут обладать дискриминирующей силой для выявления неблагоприятных контактов остаток-остаток и прояснения серьезных ограничений на возможные замены аминокислот. Следовательно, дополнительное использование структурной информации помогает правильно идентифицировать более важные остатки методом MFS.
Эффективность MFS для понимания взаимодействий белковых доменов.
MFS также можно использовать вручную, чтобы получить представление о структуре и функциях не охарактеризованных белков, что облегчает генерацию гипотез для биохимических экспериментов.Ранее мы сообщали о присутствии двух тубулиноподобных генов, бактериального тубулина a ( btuba ) и бактериального тубулина b ( btubb ) у бактерий Prosthecobacter dejongeii [52]. У эукариот α и β тубулины образуют димеры, и димеры соединяются друг с другом с образованием олигомеров, которые удлиняются с образованием протофиламентов. Протофиламенты составляют цитоскелет микротрубочек, который присутствует у всех известных эукариот, но не у бактерий или архей. Следовательно, присутствие тубулиноподобных генов btuba и btubb у видов бактерий вызвало большое любопытство в отношении их потенциальных структурных и функциональных ролей, а также их эволюционного происхождения [52].В нашей предыдущей публикации мы выполнили предсказание структуры на основе моделирования гомологии с использованием димера эукариотического α / β-тубулина в качестве шаблона. Мы проанализировали предсказанную димерную структуру с использованием показателей RAPDF и пришли к выводу, что btuba и btubb вряд ли образуют димеры в бактериях из-за структурных дестабилизирующих эффектов нескольких аминокислотных остатков в границах раздела димеров, которые различаются между btuba /. btubb и эукариотические тубулины [52].Это открытие было дополнительно подтверждено тем фактом, что данные электронной микроскопии не продемонстрировали наличие микротрубочноподобных структур у Prosthecobacter dejongeii [52]. Однако в 2005 г. кристаллические структуры butba и btubb были решены в E.coli , показывая, что btuba и btubb образуют димеры [53]. Кроме того, анализ сборки in vitro в E.coli продемонстрировал, что btuba и btubb образуют протофиламенты, содержащие равные концентрации btuba и btubb , что позволяет предположить, что две субъединицы имеют альтернативное расположение вдоль протофиламенты [54].Поэтому мы тщательно пересмотрели, почему наши предыдущие предсказания относительно образования димеров были ошибочными.
Сначала мы сравнили нашу предсказанную структуру в 2002 году с экспериментальной структурой, которая была решена в 2005 году, и обнаружили, что предсказания структуры достаточно точны: C α RMSD для btuba (433 остатка) и btubb (426 остатков). между предсказанными и экспериментальными структурами 2,28 Å и 2,36 Å соответственно. Затем мы сгенерировали метафункциональные сигнатуры для димера btuba / btubb , используя предсказанную структуру (Рисунок 4, слева).В нашем протоколе генерации MFS используется немного другая оценка структурной стабильности (RAPDF с 37 ячейками [32]), чем в предыдущей публикации, RAPDF с 18 ячейками [30]. Изучая оценки структурной стабильности интерфейса димера, мы подтвердили наши предыдущие предсказания, что димерные структуры со специфическими для бактерий заменами, такими как G100, менее стабильны [52]. Однако, исследуя 10 остатков с наивысшими показателями MFS (20 остатков, изображенных в виде красных сфер в димере) во всей структуре, мы четко различаем кластер остатков с высокими показателями, окружающий GDP на границе раздела димера.Показатели MFS подтверждают гипотезу о том, что поверхность раздела димеров действительно функционально важна и связывается с молекулами GDP, в отличие от прогнозов, порождаемых только структурной стабильностью. Этот пример дополнительно подчеркивает важность использования метафункциональных сигнатур, а не только оценок структурной стабильности при интерпретации структурных и функциональных ролей отдельных аминокислотных остатков. Другими словами, хотя была создана высокоточная модель атомного разрешения, функциональные сайты не были точно предсказаны до тех пор, пока мы не оценили информацию об эволюции и последовательности.Специфично для этой проблемы, мы находим кластеры с высокими показателями в голове btuba и хвосте btubb , косвенно предполагая, что хвост btubb может связываться с головкой btuba в другом димере. Следовательно, расчет MFS не только поддерживает образование димеров, но также и последовательное добавление димеров с образованием протофиламентов, что подтверждается биохимическими экспериментами [54].
Рис. 4. Применение MFS для понимания роли димера btuba / btubb в бактериальном роду Prosthecobacter с использованием предсказанных и экспериментальных структур.
Обе структуры окрашены посредством изображения остатков с более высокой оценкой MFS более интенсивным красным цветом, при этом 10 наиболее высоко оцениваемых остатков представлены сферами. Один GTP и один GDP в прогнозируемой структуре, а также один GDP и два иона SO 4 2- в экспериментальной структуре показаны желтыми сферами. Предсказанная структура генерируется методами моделирования гомологии с использованием димера тубулина эукариот α / β (идентификатор PDB: 1jff) в качестве шаблона. Таксоловый лиганд и ионы металлов опущены в предсказанной структуре для облегчения описания.В предсказанной структуре btubb лежит выше btubb с молекулой GDP, заключенной на границе раздела димеров. В экспериментальной структуре (идентификатор PDB: 2btq) btuba лежит выше btubb , и на границе димера GDP отсутствует. Наш анализ MFS сначала подтвердил, что btuba и btubb действительно образуют димеры из-за существования кластера с высокими показателями на границе их димеров, в отличие от предыдущих предсказаний, сделанных с использованием только показателя структурной стабильности.Кроме того, MFS предполагает, что независимо от того, как btuba и btubb ориентируются друг с другом, их интерфейс является функционально важным и может связываться с молекулами GDP.
https://doi.org/10.1371/journal.pcbi.1000181.g004
Затем мы исследовали экспериментальные структуры для димера btuba / butbb и рассчитали метафункциональные сигнатуры для димера (рис. 4, справа). Неожиданно мы обнаружили, что экспериментальная структура димера btuba / btubb отличается от нашей предсказанной структуры (а также экспериментальной структуры димера эукариотического тубулина) относительным положением димерных субъединиц.В димере эукариот, когда GDP-связывающий домен α- и β-тубулина ориентирован на север, α-тубулин располагается выше β-тубулина, так что α-тубулин связывается с GDP в нуклеотид-связывающем домене β-тубулина. Напротив, в экспериментальной структуре бактерий тубулина btuba лежит выше btubb , и на их границе раздела нет молекулы GDP, но вместо этого есть два иона SO 4 2− (показаны как два маленьких желтых сферы). Тем не менее, посредством анализа MFS мы все же обнаружили кластер высоко оцениваемых остатков на интерфейсе btuba / butbb в экспериментальных структурах, указывая на то, что этот интерфейс может быть функционально важным сайтом связывания.Принимая во внимание относительно большой промежуток между btuba и btubb на границе раздела димеров в экспериментальной структуре, существование двух ионов SO 4 2-, которые очень похожи на две фосфатные группы в GDP, и кластера высоких -счетные остатки, предложенные анализом MFS, вместе эти свидетельства указывают на сходные паттерны взаимодействия между btuba / btubb в бактериях и α / β тубулином у эукариот, несмотря на их различия в сборке, что может быть связано с артефактами кристаллографии и / или из-за к недостаточной концентрации молекул GDP в растворе.
Наконец, вычисляя метафункциональные сигнатуры для экспериментальных и предсказанных структур для btuba и btubb , мы идентифицировали несколько аминокислотных мутаций, которые дают самые высокие оценки MFS для димерной структуры. Таким образом, метафункциональные сигнатуры предполагают определенные аминокислоты, которые могут быть введены в виде мутаций в тубулины Prosthecobacter dejongeii для функциональной характеристики (в противном случае это заняло бы много часов ручного анализа).Детальные биохимические эксперименты и эксперименты по мутагенезу продолжаются для этих предсказанных важных мутаций. Этот тип подробного (и ориентированного на конкретную проблему) анализа — это то, как мы предполагаем, что MFS можно использовать для получения критического понимания роли определенных аминокислот в функции белков и для руководства экспериментальной работой.
Характеристика редких механизмов функции белков с помощью MFS.
Мы также применили MFS для характеристики механизмов белков, функции которых существенно отличаются от функций в обучающих наборах, которые ограничены каталитическими сайтами и сайтами связывания белок-лиганд.Связывание белков с биоминеральными поверхностями — это плохо изученный процесс. Одним из немногих белков млекопитающих, которые, как известно, связывают гидроксиапатитную поверхность кости, и единственный, для которого механизм был охарактеризован на атомном уровне, является остеокальцин, который, таким образом, представляет собой пример применимости MFS для прогнозирования редких механизмов функции белков.
Дифракционная структура остеокальцина (идентификатор PDB: 1q8h) [55] демонстрирует задействованные специфические остатки и иллюстрирует механизм давно известной функции связывания с поверхностью гидроксиапатита кости [56].Специфическое размещение ионов кальция вдоль внешней поверхности белка соответствует конформации ионов кальция вдоль открытой поверхности гидроксиапатита в кости [55]. Как самый распространенный неколлагеновый белок в кости [57], остеокальцин регулирует формирование кости [58], и недавно было показано, что он играет ключевую роль в эндокринной регуляции системного метаболизма [59].
Среди пяти лучших результатов SeqonlyMFS успешно идентифицировал три известных гидроксиапатит-связывающих остатка остеокальцина (17E-21E-24E), а две другие пять лучших оценок выделили два цистеина, участвующих в стабилизирующем складку дисульфидном мостике (23C – 29C; рис. 5).Полный MFS создает аналогичное распределение наивысшего функционального значения, но увеличивает оценку тирозина (42Y) над двумя остатками, связывающими гидроксиапатит. Из-за этого высокого показателя MFS мы полагаем, что 42Y представляет собой фосфорилированный остаток (а не три других тирозина), регулирующий клеточную сигнальную функцию для остеокальцина, что было показано противоположным действием на функцию поджелудочной железы у мышей, лишенных остеокальцина, по сравнению с мышами, у которых отсутствует белок. тирозинфосфатаза OST-PTP [59].Немного сниженная селективность связывающих остатков гидроксиапатита при включении показателей структурной стабильности также соответствует уменьшенному влиянию мутаций на стабильность, когда функциональные боковые цепи присутствуют вдоль свободной внешней поверхности белка, а не внутри компактной каталитической щели. Здесь строгость MFS демонстрируется сохранением всех этих остатков в первой десятке, несмотря на небольшое влияние на нестабильность. Хотя функциональные остатки в белке включают три глутаминовые кислоты, которые часто представлены как функциональные сайты в наборах данных для обучения, мы отмечаем, что MFS и SeqonlyMFS предоставляют дополнительную информацию только к информации об идентичности аминокислот: во-первых, в белке еще есть пять глутаминовых кислот. только три действительных функциональных сайта были выбраны как MFS, так и SeqonlyMFS; во-вторых, два цистеина, которые образуют дисульфидный мостик, были правильно идентифицированы как высокоэффективные остатки как MFS, так и SeqonlyMFS, однако цистеины редко представлены как функциональные сайты в наших наборах данных для обучения.Следовательно, одной лишь идентичности аминокислот недостаточно для вывода функционально важных остатков для этого белка.
Рисунок 5. Прогноз остатков с редкой функцией, не представленной в обучающих наборах.
MFS обучили на наборе остатков, экспериментально охарактеризованных для участия в канонических каталитических функциях и интерфейсах белок-лиганд. Связывание белков с биоминеральными поверхностями является редкой функцией и плохо изученным процессом, для которого единственной доступной дифракционной структурой являются ионы металла, связывающие остеокальцин (изображенные в виде зеленых сфер с ионными связями с остатками γ-карбоксиглутаминовой кислоты (gla) в прозрачной зеленой пробирке) (Идентификатор PDB: 1q8h).Три остатка gla остеокальцина (представленные в виде сфер, подобных целевым остаткам на Рисунке 3 выше), ранее показанные для связывания с гидроксиапатитной поверхностью кости, явно выбираются MFS в пределах шести верхних из 49 остатков, с или без знания структурных и посттрансляционная модификация этих остатков. Эти остатки отобраны ConSurf из восьми с гораздо меньшим различием оценок других остатков в остеокальцине. Ни один из этих остатков не выбран ET в топ-10.Этот пример демонстрирует применимость MFS для получения высокоточных и конкретных прогнозов для белков с самыми разными функциями.
https://doi.org/10.1371/journal.pcbi.1000181.g005
Для сравнения, метод Evolutionary Trace не позволяет выделить остатки, связывающие гидроксиапатит, в топ-10 баллов, оценивая их с четырнадцатого по шестнадцатый из тридцати семи. считается из конструкции. Между тем, метод ConSurf отбирает эти остатки в пределах восьми наивысших оценок, но обеспечивает гораздо более слабое различение от остального белка.Значительное падение оценок происходит после первых шести оценок в обоих распределениях MFS, в то время как более двух третей остатков оцениваются в пределах этого диапазона падения для ConSurf. Это различие в способности различать ясно видно из рисунка 5.
Функциональные α-карбоксиглутаминовые кислоты, обнаруженные методами на основе последовательностей в MFS, были упрощены во всех предсказаниях как глутаминовые кислоты в соответствии с кодирующими нуклеотидными последовательностями, так что посттрансляционная модификация не рассматривалась.Этот пример демонстрирует, что MFS можно надежно использовать для идентификации функциональных остатков, даже если структура и посттрансляционные модификации неизвестны, а остатки не участвуют в канонических каталитических реакциях или взаимодействиях белок-лиганд. Идентификация этих модифицированных остатков указывает на то, что MFS непосредственно полезен для предсказания сайтов посттрансляционной модификации. Наконец, структурное упрощение, используемое в нашем анализе, объясняет более слабое различение этих функциональных остатков при рассмотрении структуры, поскольку экспериментальная структура дестабилизируется из-за увеличенного объема и отрицательного заряда боковых цепей α-карбоксиглутаминовой кислоты.
Уточнение выравниваний для сравнительного моделирования.
Мы исследовали использование MFS для помощи в создании парных выравниваний для отдаленно родственных белков. Создание точных парных выравниваний важно для предсказания структуры белка с использованием методов моделирования гомологии, потому что обычно первым шагом в моделировании гомологии является копирование атомных координат целевого белка в запрашиваемый белок для всех выровненных остатков. Некоторые из лучших выравниваний производятся сервером 3D-Jury [60], который представляет собой мета-сервер, который собирает информацию о выравнивании, а также оценивает информацию от многих отдельных серверов для выравнивания структуры последовательности и генерирует согласованное попарное выравнивание.Один из белков, над которым мы работали, — это белок dtx с 639 остатками. Мы отправили последовательность запросов на сервер 3D-Jury, чтобы определить экспериментальную структуру, которая имеет наилучшую оценку соответствия запросу. Мы использовали выравнивание с наивысшей оценкой 3D-жюри для структурного моделирования. Один сегмент созданного нами попарного выравнивания:
Запрос: GIREHAMGAIMNGISAFGANYKPYGGTFLNFVSYA
Цель: GIAEQHAMTSAAGLAMGG – LHPVVAIYSTFLNRA
Однако, когда мы вычисляем SeqonlyMFS как для запрашиваемого белка, так и для целевого белка, мы обнаружили, что как «H» (гистидины) в запросе, так и целевая последовательность входят в десятку наиболее высоко оцениваемых остатков.Эта функциональная сигнатура предполагает, что может быть ошибка выравнивания; следовательно, лучшее согласование, основанное на функциональных доказательствах:
Запрос: GIRE-HAMGAIMNGISAFGANYKPYGGTFLNFVSYA
Цель: GIAEQHAMTSAAGLAMGG — LHPVVAIYSTFLNRA
, который вводит два дополнительных пробела (обычно нежелательных для моделирования структуры), но выравнивает функционально важные остатки друг с другом. Наличие более точных трехмерных координат для функционально важных остатков и областей будет особенно важно в последующем функциональном анализе и генерации гипотез для предсказанных структур.Поскольку экспериментальная структура для этого белка еще не доступна, мы не смогли дополнительно подтвердить точность MFS-скорректированных выравниваний со структурной точки зрения. Вышеупомянутая процедура является просто примером ручной корректировки парных выравниваний для удаленно связанных белков; однако при более сложной разработке алгоритмов станет возможным генерировать функциональные выравнивания, в отличие от выравниваний последовательностей или структур, в автоматическом режиме для двух белков с функциями, которые представлены несколькими векторами переменной длины.В таких случаях, вместо предсказания функциональных остатков, MFS-подобная процедура может использоваться для переноса аннотации между двумя белками. Фактически, ключевые функциональные особенности белковых структур уже использовались для улучшения передачи аннотаций между ферментами [61]. Такие функциональные выравнивания были бы полезны как для предсказания структуры, так и для функциональных исследований неохарактеризованных белков.
Обсуждение
В этой работе мы описываем протокол генерации метафункциональной сигнатуры (MFS), который объединяет несколько источников информации для предсказания функционального сайта белка.Мы также демонстрируем способность этого протокола характеризовать функцию белка на основе остатков, используя четыре реальных примера.
Ключевые идеи, представленные в этом исследовании, включают разделение структурного и функционального вкладов, использование псевдоэнергетических функций для мутированных структур для определения их влияния на функцию белка, а также сочетание основанных на знаниях и биофизических подходов для всестороннего аннотирования функциональное значение остатков в последовательности белка.Большинство компонентов нашего подхода не уникальны: другие алгоритмы прогнозирования функций используют множественное выравнивание последовательностей, информацию из базы данных, а также экспериментальные и предсказанные структуры белков. Одним из уникальных аспектов нашего подхода является интеграция всех компонентов в единую структуру, основанную на знаниях и структуре, которая может обеспечивать более точные и полные прогнозы, но каждый компонент также может обеспечивать различные аспекты биологического понимания интерпретации белка. функция.
Поскольку в нашем исследовании использовались два разных набора данных (набор Торнтона и набор Ловелла) из разных источников, мы хотим сравнить и обсудить здесь параметры модели для разных наборов данных. Этот анализ может помочь нам понять относительный вклад различных компонентов оценки в двух наборах данных. Чтобы учесть различную величину переменных-предикторов, мы вычислили наклон коэффициента регрессии при преобразовании всех предикторов в Z-баллы.Для набора данных Thornton наклон для нормализованных HMM_rel_ent, SSR, RAPDF_spread и RAPDF_dif составляет 1,1, 0,25, 0,52 и 0,23 соответственно; для набора данных Lovell соответствующие значения равны 1,1, 0,28, 0,45 и 0,19 соответственно. Следовательно, для набора данных Thornton, который содержит каталитические сайты, модель содержит немного больший вклад от оценок на основе структуры, что указывает на то, что структурная информация относительно более важна для определения каталитических сайтов, чем интерфейсы связывания.Кроме того, мы также сравнили относительный вклад 20 аминокислот в модель. Для набора данных Thornton пять аминокислот с наибольшим вкладом — это Glu, Lys, Asp, Arg и Ser, соответственно, с нормализованными коэффициентами в диапазоне от 0,55 до 0,83. Для набора данных Ловелла пять аминокислот с наибольшим вкладом также являются Glu, Lys, Asp, Arg и Ser, соответственно, с нормализованными коэффициентами в диапазоне от 0,66 до 0,84. Следовательно, идентичность аминокислот, по-видимому, играет одинаково важную роль в этих двух наборах данных.Мы отмечаем, что «функциональные остатки» в контексте этого исследования представляют собой как каталитические сайты, так и сайты связывания, однако из-за ограничений источников данных каждый набор тестовых данных содержит только часть истинных функциональных сайтов, поэтому некоторые истинно положительные результаты могут быть неправильно обрабатываются как нефункциональные сайты в каждом наборе данных. Помимо сравнения двух наборов данных, чтобы оценить стабильность регрессионных моделей, мы также выполнили аналогичный анализ, сравнив пять наборов моделей, используемых в экспериментах с перекрестной проверкой, и обнаружили, что параметры модели в основном идентичны между перекрестными проверками (данные не показано).
Хотя мы представили MFS как набор компонентов скоринга, интегрированных простой моделью логистической регрессии, альтернативным способом интеграции информации является использование сложного подхода машинного обучения, например, с помощью алгоритмов на основе SVM. Мы исследовали эту проблему, но решили использовать регрессионную модель по нескольким причинам: во-первых, хотя хорошо известно, что SVM хорошо работает с задачами бинарной классификации, она страдает отсутствием «биологической» интерпретации. Например, Петрова и др. Оценили 26 различных алгоритмов / классификаторов в программном пакете WEKA и представили лучшую комбинацию компонентов в виде набора семи (из 24) свойств остатков для прогнозирования каталитических остатков [37].Кроме того, Юн и др. Протестировали SVM на 314 различных функциях, продемонстрировали, что совместное использование нескольких функций улучшает производительность, и представили функции с наиболее высоким рейтингом [39]. Pugalenthi et al. протестировали 278 различных функций для предсказания каталитических сайтов и исследовали производительность при использовании подмножества из 50–250 функций [40]. Хотя эти подходы к машинному обучению обычно приводят к повышению производительности, трудно декодировать эти методы «черного ящика» и использовать отдельный компонент (из десятков или сотен) для интерпретации различных аспектов биологической функции, как мы это сделали с MFS на четыре реальных примера.Следовательно, в этих случаях простая модель логистической регрессии является концептуально лучшим выбором, где параметры регрессии легко понятны. Во-вторых, функциональная важность может быть эффективно отражена несколькими в значительной степени независимыми характеристиками простой линейной модели, не прибегая к тестированию многих более сложных моделей и выбору наиболее эффективной модели. Например, на рисунке 1 Петровой и др., Хотя SVM занимает более высокое место, чем логистическая регрессия при сравнении множества различных алгоритмов, производительность этих двух методов действительно очень схожа.Поэтому мы полагались на простую модель логистической регрессии как на лучший подход для представления и интеграции ансамбля методов, основанных на знаниях и биофизике, в MFS.
Больше, чем просто другой алгоритм прогнозирования функционального сайта, MFS может использоваться как способ определения функции белка с помощью ряда количественных значений, которые отражают функциональную важность белка. Абстрагируя функцию белка в вектор (или несколько векторов, если каждый отдельный компонент рассматривается отдельно), можно применять более сложные алгоритмы для более эффективного использования этой информации.Традиционно два белка могут быть выровнены вместе на основании сходства их последовательностей, сходства структур или совместимости последовательности-структуры. Однако введение концепции MFS позволяет генерировать функциональное выравнивание между двумя белками. Например, мы продемонстрировали, что, сравнивая оценки MFS для двух белков, мы потенциально можем улучшить точность выравнивания, используя функциональные сигнатуры вручную. Однако автоматический алгоритм выравнивания двух матриц переменной длины нетривиален.Необходимы усовершенствования алгоритмов, чтобы найти оптимальное решение для автоматического функционального выравнивания двух белков. Мы активно ищем приблизительные решения этой проблемы.
Помимо методов идентификации функциональных сайтов, используемых в статье, мы понимаем, что существует множество других различных типов методов для идентификации важных остатков из последовательности или структуры белка. Многие из методов основаны на непрерывной последовательности аминокислотных паттернов, например паттерн PROSITE [62] и паттерн BLOCKs [63].Все остатки в данном белке, которые соответствуют определенным мотивам, считаются функционально важными, и свойства мотивов также могут указывать на определенные функциональные роли белка. Однако эти методы обычно приводят к значительному завышенному предсказанию остатков «функционального сайта»; например, некоторые паттерны PROSITE состоят из мотивов с 3 остатками, которые соответствуют множеству сайтов в множестве белков. Следовательно, хотя эти методы полезны для подтверждения существования паттерна, соответствующего биологической функции, или для генерации гипотез для прогнозирования возможной функциональной категории, эти методы обычно слишком общие для определения функциональной важности на уровне остатков.Мы рассматриваем наш метод и методы сканирования мотивов как идеологически разные методологии решения схожих задач. Вместе они могут помочь пользователям получить дополнительную биологическую информацию для характеристики белков.
Протокол генерации MFS можно улучшить несколькими способами. Одним из преимуществ концепции MFS является то, что она состоит из нескольких независимых модулей, поэтому каждый модуль можно обновлять и улучшать без нарушения функциональности других модулей. Мы улучшаем производительность MFS во многих аспектах.Во-первых, хотя многие другие веб-серверы (например, SIFT) используют весь NR или всю коллекцию последовательностей TrEMBL, мы использовали только данные Uniref90, что позволило нам ускорить поиск BLAST. Однако набор данных Uniref90 не самого высокого качества. Существует много очень коротких последовательностей, которые можно легко включить в выравнивания, а многие неизвестные аминокислоты аннотированы как длинные отрезки «X». Кроме того, мы использовали программу PSI-BLAST для сканирования базы данных последовательностей и генерации множественных выравниваний, которые на самом деле представляют собой просто накопленную версию множественных попарных выравниваний.Создание более точных множественных выравниваний поможет при оценке сохранности на основе последовательностей и выводах о филогении. Кроме того, расчет RAPDF для мутированных структур также может быть оптимизирован. Необязательный шаг после замены боковой цепи — минимизация энергии за счет глобального возмущения структуры. Этот шаг может быть реализован с помощью протокола ENCAD [48]. Поскольку эта процедура значительно увеличивает время выполнения, мы сделали ее необязательным шагом. Более быстрое создание более точных оценок структурной стабильности для мутировавших структур улучшило бы производительность MFS.Дальнейшая разработка и оптимизация текущего протокола значительно улучшат функциональную аннотацию пространства последовательностей и структур.
Помимо повышения эффективности прогнозирования функционального сайта белка, оценки MFS, рассматриваемые как векторы, могут использоваться для различения функциональных категорий для данного белка (например, назначение суперсемейства SCOP [35], [64] или узла GO в иерархии GO ). Анализ MFS также выявляет функциональную важность на уровне остатков, что позволяет проводить рациональный мутагенез и биохимические эксперименты.Наконец, метод MFS может быть использован для изменения функции белка, что приведет к его применению в дизайне белков и открытии лекарств. Применение протоколов MFS во многих областях вычислительной биологии и биоинформатики, как показано на примерах в статье, может значительно продвинуть наше понимание взаимосвязи белковой последовательности, структуры и функции и направить экспериментальную характеристику функции белка.
Благодарности
Мы благодарим Рене Иретон за критическое прочтение и редактирование рукописи.Мы также хотим поблагодарить членов группы вычислительной биологии Samudrala за полезные обсуждения и комментарии.
Вклад авторов
Задумал и спроектировал эксперименты: RS. Проведенные эксперименты: KW. Проанализированы данные: KW JAH. Предоставленные реагенты / материалы / инструменты анализа: GC DCN. Написал статью: KW.
Ссылки
- 1.
Watson JD, Laskowski RA, Thornton JM (2005) Предсказание функции белка на основе данных последовательности и структуры.Curr Opin Struct Biol 15: 275–284. - 2.
Whisstock JC, Lesk AM (2003) Прогнозирование функции белка на основе последовательности и структуры белка. Q Rev Biophys 36: 307–340. - 3.
Фридберг I (2006) Автоматизированное предсказание функции белков — геномная проблема. Краткая биография 7: 225–242. - 4.
Webb EC (1992) Enzyme Nomenclature 1992. Сан-Диего (Калифорния): Academic Press. - 5.
Эшбернер М., Болл К.А., Блейк Дж. А., Ботштейн Д., Батлер Н. и др.(2000) Генная онтология: инструмент для объединения биологии. Нат Генет 25: 25–29. - 6.
Андреева А., Ховорт Д., Бреннер С.Е., Хаббард Т.Дж., Чотия С.и др. (2004) База данных SCOP в 2004 году: уточнения объединяют данные о структуре и семействе последовательностей. Нуклеиновые кислоты Res 32: D226 – D229. - 7.
Валдар В.С. (2002) Сохранение остатков в баллах. Белки 48: 227–241. - 8.
Sander C, Schneider R (1991) База данных гомологичных белковых структур и структурное значение выравнивания последовательностей.Белки 9: 56–68. - 9.
Шенкин П.С., Эрман Б., Мастрандреа Л.Д. (1991) Теоретико-информационная энтропия как мера изменчивости последовательности. Белки 11: 297–313. - 10.
Williamson RM (1995) Информационно-теоретический анализ взаимосвязи между структурой первичной последовательности и узнаванием лиганда среди класса облегченных переносчиков. Дж. Теор Биол 174: 179–188. - 11.
Мирный Л.А., Шахнович Е.И. (1999) Универсально консервативные позиции в белковых складках: считывание эволюционных сигналов о стабильности, кинетике складывания и функции.J Mol Biol 291: 177–196. - 12.
Plaxco KW, Larson S, Ruczinski I., Riddle DS, Thayer EC, et al. (2000) Эволюционная консервация кинетики сворачивания белков. J Mol Biol 298: 303–312. - 13.
Герштейн М., Альтман Р. Б. (1995) Средние основные структуры и меры вариабельности для семейств белков: применение к иммуноглобулинам. J Mol Biol 251: 161–175. - 14.
Пей Дж., Дохолян Н.В., Шахнович Е.И., Гришин Н.В. (2003) Использование дизайна белков для обнаружения гомологии и поиска активных сайтов.Proc Natl Acad Sci U S A 100: 11361–11366. - 15.
Валдар В.С., Торнтон Дж. М. (2001) Интерфейсы белок-белок: анализ консервации аминокислот в гомодимерах. Белки 42: 108–124. - 16.
Гривз Р., Уорвикер Дж. (2005) Идентификация активного сайта с помощью расчетов на основе геометрии и профиля последовательности: захоронение каталитических щелей. J Mol Biol 349: 547–557. - 17.
Пей Дж., Гришин Н.В. (2001) AL2CO: расчет позиционной консервативности при выравнивании белковой последовательности.Биоинформатика 17: 700–712. - 18.
Capra JA, Singh M (2007) Предсказание функционально важных остатков на основе сохранения последовательности. Биоинформатика 23: 1875–1882. - 19.
Ван К., Самудрала Р. (2006) Включение фоновой частоты улучшает меры по сохранению остатков на основе энтропии. BMC Bioinformatics 7: 385. - 20.
Lichtarge O, Bourne HR, Cohen FE (1996) Метод эволюционного отслеживания определяет поверхности связывания, общие для семейств белков.J Mol Biol 257: 342–358. - 21.
Яо Х., Кристенсен Д.М., Михалек И., Сова М.Э., Шоу С. и др. (2003) Точный, чувствительный и масштабируемый метод определения функциональных сайтов в белковых структурах. J Mol Biol 326: 255–261. - 22.
Mihalek I, Res I, Lichtarge O (2006) Evolutionary trace report_maker: новый тип сервиса для сравнительного анализа белков. Биоинформатика 22: 1656–1657. - 23.
Mihalek I, Res I, Lichtarge O (2004) Семейство эволюционно-энтропийных гибридных методов для ранжирования белковых остатков по важности.J Mol Biol 336: 1265–1282. - 24.
Ландау М., Мэйроуз I, Розенберг Й., Глейзер Ф., Марц Э. и др. (2005) ConSurf 2005: проекция эволюционной оценки сохранности остатков в белковых структурах. Нуклеиновые кислоты Res 33: W299 – W302. - 25.
Глейзер Ф., Пупко Т., Паз I, Белл Р.Э., Бехор-Шентал Д. и др. (2003) ConSurf: идентификация функциональных областей в белках путем картирования поверхности филогенетической информации. Биоинформатика 19: 163–164. - 26.Soyer OS, Goldstein RA (2004) Предсказание функциональных сайтов в белках: сайт-специфические эволюционные модели и их применение к транспортерам нейротрансмиттеров. J Mol Biol 339: 227–242.
- 27.
La D, Sutch B, Livesay DR (2005) Предсказание функциональных сайтов белков с филогенетическими мотивами. Белки 58: 309–320. - 28.
del Sol Mesa A, Pazos F, Valencia A (2003) Автоматические методы прогнозирования функционально важных остатков. J Mol Biol 326: 1289–1302. - 29.
Dessailly BH, Lensink MF, Wodak SJ (2007) Связывание дестабилизирующих областей с известными функциональными сайтами в белках. BMC Bioinformatics 8: 141. - 30.
Samudrala R, Moult J (1998) Всеатомная зависимая от расстояния условная дискриминирующая функция вероятности для предсказания структуры белка. J Mol Biol 275: 895–916. - 31.
Ван К., Фейн Б., Левитт М., Самудрала Р. (2004) Улучшенный выбор структуры белка с использованием зависимых от приманки дискриминационных функций.BMC Struct Biol 4: 8. - 32.
Лю Т., Самудрала Р. (2006) Влияние экспериментального разрешения на выполнение основанных на знаниях дискриминационных функций для выбора структуры белка. Protein Eng Des Sel 19: 431–437. - 33.
Hung LH, Ngan SC, Liu T, Samudrala R (2005) PROTINFO: новые алгоритмы для улучшенного предсказания структуры белка. Нуклеиновые кислоты Res 33: W77-W80. - 34.
Chelliah V, Chen L, Blundell TL, Lovell SC (2004) Выявление структурных и функциональных ограничений в эволюции с целью выявления мест взаимодействия.J Mol Biol 342: 1487–1504. - 35.
Ван К., Самудрала Р. (2005) FSSA: новый метод определения функциональных сигнатур по структурным согласованиям. Биоинформатика 21: 2969–2977. - 36.
Cheng G, Qian B, Samudrala R, Baker D (2005) Улучшение предсказания функционального сайта белка путем выделения структурных и функциональных ограничений на эволюцию семейства белков с использованием вычислительного дизайна. Nucleic Acids Res 33: 5861–5867. - 37.
Петрова Н.В., Ву СН (2006) Прогнозирование каталитических остатков с помощью машины опорных векторов с выбранной последовательностью белка и структурными свойствами.BMC Bioinformatics 7: 312. - 38.
Fischer JD, Mayer CE, Soding J (2008) Прогнозирование функциональных остатков белка на основе последовательности путем оценки плотности вероятности. Биоинформатика 24: 613–620. - 39.
Youn E, Peters B, Radivojac P, Mooney SD (2007) Оценка характеристик для предсказания каталитических остатков в новых складках. Protein Sci 16: 216–226. - 40.
Pugalenthi G, Kumar KK, Suganthan PN, Gangal R (2008) Идентификация каталитических остатков из структуры белка с использованием машины опорных векторов с последовательностью и структурными особенностями.Biochem Biophys Res. Commun. 367: 630–634. - 41.
Tang YR, Sheng ZY, Chen YZ, Zhang Z (2008) Улучшенное предсказание каталитических остатков в структурах ферментов. Protein Eng Des Sel 21: 295–302. - 42.
Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, et al. (2006) Универсальный белковый ресурс (UniProt): расширяющаяся вселенная информации о белках. Нуклеиновые кислоты Res 34: D187 – D191. - 43.
Альтшул С.Ф., Мэдден Т.Л., Шаффер А.А., Чжан Дж., Чжан З. и др.(1997) Gapped BLAST и PSI-BLAST: новое поколение программ поиска в базе данных белков. Nucleic Acids Res 25: 3389–3402. - 44.
Eddy SR (1998) Профиль скрытых марковских моделей. Биоинформатика 14: 755–763. - 45.
ФИЛИП http://evolution.genetics.washington.edu/phylip.html. - 46.
Самудрала Р., Левитт М. (2002) Всесторонний анализ 40 слепых предсказаний структуры белка. BMC Struct Biol 2: 3. - 47.
Канутеску А.А., Шеленков А.А., Данбрак Р.Л. мл. (2003) Алгоритм теории графов для быстрого предсказания боковой цепи белка.Protein Sci 12: 2001–2014. - 48.
Левитт М., Хиршберг М., Шарон Р., Даггетт В. (1995) Функция и параметры потенциальной энергии для моделирования молекулярной динамики белков и нуклеиновых кислот в растворе. Comput Phys Commun 91: 215–231. - 49.
Райс П., Лонгден И., Близби А. (2000) EMBOSS: Открытый программный пакет европейской молекулярной биологии. Тенденции Genet 16: 276–277. - 50.
Porter CT, Bartlett GJ, Thornton JM (2004) Атлас каталитических сайтов: ресурс каталитических сайтов и остатков, идентифицированных в ферментах с использованием структурных данных.Нуклеиновые кислоты Res 32: D129 – D133. - 51.
Петтерсен Э.Ф., Годдард Т.Д., Хуанг С.С., Коуч Г.С., Гринблатт Д.М. и др. (2004) UCSF Chimera — система визуализации для поисковых исследований и анализа. J Comput Chem 25: 1605–1612. - 52.
Дженкинс С., Самудрала Р., Андерсон И., Хедлунд Б.П., Петрони Дж. И др. (2002) Гены цитоскелетного белка тубулина у бактерий рода Prosthecobacter. Proc Natl Acad Sci U S A 99: 17049–17054. - 53.
Schlieper D, Oliva MA, Andreu JM, Lowe J (2005) Структура бактериального тубулина BtubA / B: доказательства горизонтального переноса генов.Proc Natl Acad Sci U S A 102: 9170–9175. - 54.
Sontag CA, Staley JT, Erickson HP (2005) Сборка in vitro и гидролиз GTP бактериальными тубулинами BtubA и BtubB. J Cell Biol 169: 233–238. - 55.
Hoang QQ, Sicheri F, Howard AJ, Yang DS (2003) Механизм распознавания костей остеокальцина свиньи по кристаллической структуре. Природа 425: 977–980. - 56.
Poser JW, Price PA (1979) Метод декарбоксилирования γ-карбоксиглутаминовой кислоты в белках.Свойства декарбоксилированного белка γ-карбоксиглутаминовой кислоты из костей теленка. J Biol Chem 254: 431–436. - 57.
Hauschka PV, Lian JB, Cole DE, Gundberg CM (1989) Остеокальцин и матриксный белок Gla: витамин К-зависимые белки в кости. Physiol Rev 69: 990–1047. - 58.
Ducy P, Desbois C, Boyce B, Pinero G, Story B и др. (1996) Повышенное костеобразование у мышей с дефицитом остеокальцина. Nature 382: 448–452. - 59.
Ли Н.К., Сова Х., Хинои Э., Феррон М., Ан Дж. Д. и др.(2007) Эндокринная регуляция энергетического обмена скелетом. Cell 130: 456–469. - 60.
Гинальски К., Элофссон А., Фишер Д., Рыхлевски Л. (2003) 3D-Jury: простой подход для улучшения прогнозов структуры белка. Биоинформатика 19: 1015–1018. - 61.
Уорд Р.М., Эрдин С., Тран Т.А., Кристенсен Д.М., Лисевски А.М. и др. (2008) Деорфанизация структурного протеома путем взаимного сравнения эволюционно важных структурных особенностей. PLoS ONE 3: e2136. - 62.
Хуло Н., Байрох А., Буллиард В., Серутти Л., Де Кастро Е. и др. (2006) База данных PROSITE. Нуклеиновые кислоты Res 34: D227 – D230. - 63.
Ng PC, Henikoff S (2003) SIFT: прогнозирование аминокислотных изменений, влияющих на функцию белка. Nucleic Acids Res 31: 3812–3814. - 64.
Ван К., Самудрала Р. (2006) Автоматическая функциональная классификация экспериментальных и прогнозируемых белковых структур. BMC Bioinformatics 7: 278.
The Business of Alt Protein: интеллектуальная собственность для компаний, занимающихся альтернативными белками
Описание события
Технологические достижения в области производства мяса из растений и выращиваемых культур обещают смягчить воздействие нашей продовольственной системы на окружающую среду, снизить риск зоонозных заболеваний и, в конечном итоге, накормить больше людей с меньшими ресурсами.Этот веб-семинар от Good Food Institute и Haynes and Boone, LLP предоставит обзор того, как стратегически обеспечить защиту интеллектуальной собственности (например, патенты, коммерческую тайну и т. Д.) Для инноваций в этой сфере. Партнеры Хейнса и Буна Роджер Куан, Джейсон Новак, Бен Пеллетье и Роджер Ройз поделятся своим мнением о том, как развивающиеся компании могут лучше всего позиционировать себя для достижения успеха — от выбора правильной корпоративной структуры до подхода к привлечению капитала из венчурных и частных инвестиционных компаний.
Темы включают:
- Обзор типов защиты интеллектуальной собственности
- Когда использовать патенты по сравнению с коммерческой тайной
- Как выбрать правильную корпоративную структуру
- Подготовка к финансированию
- Планирование выхода
- Вопросы и ответы аудитории.
Динамики
Роджер Куан — партнер Haynes and Boone и председатель Группы практики точной медицины и цифрового здравоохранения, где он консультирует компании, занимающие уникальное положение в области конвергенции биологических / медицинских наук и технологических отраслей, о том, как успешно ориентироваться в сложности интеллектуальной собственности (ИС), прав на данные и нормативные проблемы, с которыми они сталкиваются.
Роджер имеет обширный опыт в области стратегии интеллектуальной собственности и управления портфелем (патенты на полезные модели / образцы, товарные знаки, авторские права и внешний вид), стратегии прав на данные, лицензирования и технологических транзакций, разрешений на свободу действий, правоприменения, монетизации, комплексной проверки интеллектуальной собственности и разрешение споров. Его практика сосредоточена в секторе наук о жизни (например, исследовательские инструменты, аналитическое оборудование / программное обеспечение, цифровая терапия, медицинские устройства, диагностика, оборудование для биопроизводства и т. Д.) с упором на новые технологии, такие как прецизионная медицина (например, платформы геномного секвенирования, AI / ML, вычислительная геномика / биоинформатика, молекулярная диагностика, сопутствующая диагностика и т. д.), цифровое здоровье (например, мобильные приложения, поддержка клинических решений, программное обеспечение , цифровая терапия, диагностика изображений AI / ML, носимые устройства и т. д.) и 3D-печать / биопечать.
Джейсон Новак — партнер в офисе Haynes and Boone в Сан-Франциско, где он специализируется на консультировании крупных и малых предприятий по различным юридическим вопросам, которые могут возникнуть в связи с новыми технологиями в пищевой промышленности, здравоохранении и биологических науках.Продукты питания и биотехнологии, а также технологии и биотехнологии — это традиционно разрозненные технологии, которые, будучи объединены вместе, чтобы сформировать многие из наших самых захватывающих новых технологий, порождают сочетание уникальных и взаимосвязанных правовых вопросов.
Джейсон имеет обширный опыт в области стратегии интеллектуальной собственности и управления патентным портфелем, подготовки и судебного преследования, возражений, консультирования, лицензирования и технологических транзакций, разработки программ выдачи и выдачи лицензий, свободы действий, различных видов должной осмотрительности, обучения в области интеллектуальной собственности и признание рисков и управление ими.Он передает этот опыт клиентам в различных отраслях, особенно в пищевой, медицинской, персонализированной / точной медицине, цифровом здравоохранении и производстве инструментов для наук о жизни.
Бен Пеллетье — партнер Группы по прецизионной медицине и цифровому здоровью в офисе Хейнса и Буна в Сан-Франциско.
Его практика сосредоточена на консультировании клиентов в сложных и быстро развивающихся областях биологии, медицины, инженерии и науки о данных.Бен имеет значительный опыт создания портфелей патентов, касающихся моноклональных антител и конъюгатов антитело-лекарство (ADC), CAR-T-клеток, стволовых клеток, тканевой инженерии, трехмерной биопечати, биосенсоров, молекулярной биологии и рабочих процессов обработки клеток, микрофлюидики, геномного секвенирования и биоинформатики. , а также сопутствующие диагностические пробирные системы и устройства.
Роджер Ройс — корпоративный и налоговый партнер в офисе юридической фирмы AmLaw 100 Haynes & Boone, LLP в Пало-Альто.Роджер является супер-юристом Северной Калифорнии, имеет экспертный рейтинг AV Мартиндейл Хаббелл и имеет оценку «Превосходно» от Avvo. Роджер также является организатором AgTech конференций Кремниевой долины и Кремниевой долины. Роджера цитировали в Wall Street Journal, Forbes, Fox Business, Chicago Tribune, Associated Press, Tax Notes, Inc.